Materials
All chemicals were purchased from Merck (Soeborg, Denmark) unless otherwise stated.
Cell culture
The immortalized human cerebral microvascular endothelial cell line (hCMEC/D3) was maintained in T75-flasks coated with collagen type I from calf skin (C9791, 18.7 µg/mL) and cultured in EBM®-2 medium (Lonza, Basel, Schweiz) supplemented with 5% (v/v) fetal bovine serum (FBS, 10270, Life Technologies, CA, USA or SH3008803, ThermoFisher Scientific, MA, USA), 100 U/mL penicillin and 100 µg/ml streptomycin, 1 ng/mL human basic fibroblast growth factor (bFGF), 1.4 µM hydrocortisone, 1% (v/v) chemically defined lipid concentrate (ThermoFisher Scientific, MA, USA), 10 mM N-2-hydroxyethylpiperazine-N-2-ethane sulfonic acid (HEPES) and 5 µg/mL ascorbic acid (5% CO2, 37°C). Culture media was changed every second or third day. Cells were passaged using trypsin-EDTA solution 10x (T4174, < 5 minutes, 5% CO2, 37°C). For experiments, cells were seeded into 24-well plates (6.7·104 cells/cm2) and cultured for four continuous days. The cells were used for experiments between passage 3 to 31.
The porcine ileum epithelial cell line IPEC-J2 transfected with human MDR1 (38) was maintained in T175-flasks and cultured in Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12, supplemented with 10% (v/v) FBS (SH3008803, ThermoFisher Scientific, MA, USA), 100 U/mL penicillin and 100 µg/mL streptomycin, 2 mM L-glutamine, and 1 mM sodium pyruvate. The culture medium was supplemented with 2 µg/mL puromycin during maintenance. Culture media was changed every second or third day. Cells were passaged using trypsin-EDTA solution 10x (5–10 minutes, 5% CO2, 37°C). For experiments, cells were either seeded into 24-well plates (1·105 cells/cm2) and cultured for 2 days or seeded on Transwell® polyester inserts (3460, 1.12 cm2, pore size 0.4 µm) (3.57·104 cells/cm2) and cultured for 15–17 days. IPEC-J2 MDR1 cells were used for experiments between passage 2 to 4.
Cellular uptake studies
The cells were washed twice with 37°C Hank’s Balanced Salt Solution (HBSS) supplemented with 10 mM HEPES, 0.0375% (v/v) sodium bicarbonate and 0.05% (w/v) bovine serum albumin (BSA) and adjusted to pH 7.4 (hHBSS). The cells were equilibrated in hHBSS with or without inhibitor for 15 minutes (37°C, 90 rpm). The compound of interest was spiked into the hHBSS followed by incubation for a designated time. The timeframe of the initial uptake rate was validated into hCMEC/D3 cells (Fig. 1 and Additional file 1). The experiment was terminated by washing three times with ice-cold HBSS supplemented with 0.0375% (v/v) sodium bicarbonate.
To assess the concentration-dependent uptake, the uptake flux was fitted to the Michaelis-Menten equation, Eq. 1.
$${\text{J}}_{\text{s}\text{s}}=\frac{{\text{J}}_{\text{m}\text{a}\text{x}} \left[\text{S}\right]}{{\text{K}}_{\text{m}}+\left[\text{S}\right]}$$
1
where Jss represents flux under steady state, Jmax is the maximal flux, [S] is the substrate concentration, and Km is the Michaelis-Menten constant.
The inhibitory constant at 50% inhibition (IC50) was determined by fitting the uptake flux (Jss) as function of the logarithmic inhibitor concentration to Eq. 2.
$${\text{J}}_{\text{s}\text{s}}={\text{J}}_{\text{m}\text{i}\text{n}}+\frac{({\text{J}}_{\text{m}\text{a}\text{x}}-{\text{J}}_{\text{m}\text{i}\text{n}})}{1+{10}^{\left(\text{log}\left(\text{I}{\text{C}}_{50}\right)-\left[\text{I}\right]\right){\text{n}}_{\text{H}}}}$$
2
where [I] is the inhibitor concentration, nH is the Hill coefficient.
Manipulation of intra- and extracellular pH
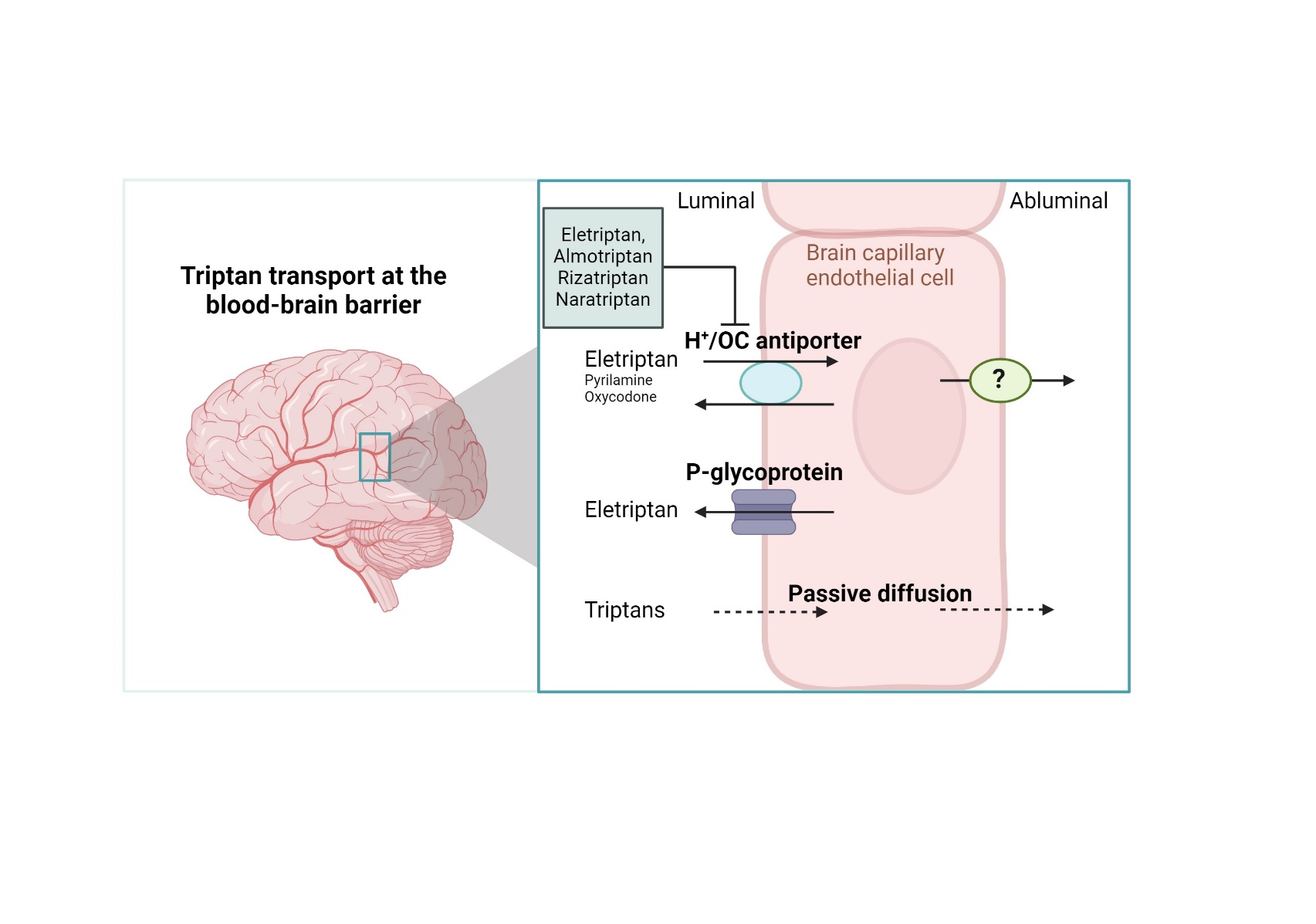
Intracellular pH was manipulated as earlier described by Byron et al. (39) to assess the involvement of a proton antiporter, i.e. the H+/OC antiporter. Briefly, elevated intracellular pH during cellular uptake was achieved by adding the substrate of interests immediately (< 10 seconds) after exposure to 30 mM NH4Cl in hHBSS. Decreased intracellular pH during cellular uptake was achieved by preincubation with 30 mM NH4Cl in hHBSS (30 minutes, 37°C) followed by aspiration and addition of hHBSS buffer. After approximately 60 seconds, the compound of interest was added to the buffer. Extracellular pH manipulation was achieved by adding hHBSS adjusted to pH 6.8, 7.4, or 8.2 during cellular uptake.
Intracellular pH measurements
Intracellular pH changes were assessed with the fluorescent pH indicator 2ʹ,7ʹ- bis-(Carboxyethyl)-5(6ʹ)-carboxyfluorescein acetoxymethyl ester (BCECF-AM) using a NOVOstar plate reader (BMG TABTECH GmbH, Offenburg, Germany). Fluorescence intensities were measured using a dual excitation of 485 and 380 nm and an emission filter of 520 nm. Briefly, hCMEC/D3 cells were seeded into a 96-well plate (3·104 cells/cm2) and cultured for three continuous days (5% CO2, 37°C). Cell media was changed the day before an experiment. A 5 µM BCECF-AM loading solution was prepared in hHBSS without BSA (hHBSS(-)) followed by sonication using a tip sonicator S-4000 (60 amplitude, 120 seconds, Misonix, NY, USA), to boost dissolution, and equilibrated to 37°C. The cells were washed twice with 37°C hHBSS(-) and loaded with the BCECF-AM solution (45 minutes, protected from light, 37°C). The cells were washed three times with 37°C hHBSS(-) and loaded with either hHBSS(-) or 30 mM NH4Cl in hHBSS(-). Control cells were preincubated with hHBSS(-) followed by automated injection of hHBSS(-) to validate the influence of injection on the fluorescence signal. Cells exposed to acute NH4Cl were preincubated with hHBSS(-) followed by automated injection of NH4Cl to a final concentration of 30 mM. For automated injections during measurements, the injection volume was 33 µL with a speed of 310 µL/s. Cells exposed to NH4Cl preincubation were preincubated with 30 mM NH4Cl in hHBSS(-) for 30–60 minutes followed by measurements of the fluorescent baseline. The NH4Cl solution was manually aspirated followed by manual loading of 100 µL hHBSS(-). Immediately thereafter, the fluorescence was assessed. The fluorescent signal was normalized against baseline signals.
Barrier integrity measurements
The barrier integrity of the IPEC-J2 MDR1 cell monolayers cultured on semipermeable supports was assessed by transepithelial electrical resistance (TEER) measurements. Cells were allowed to equilibrate at room temperature for 20 minutes. The resistance was measured using an Endohm 12-cup electrode chamber (World Precision Instruments Inc., FL, USA) connected to an EVOM voltmeter (World Precision Instruments Inc., FL, USA). Measured TEER values were subtracted by the TEER value of a blank filter (14 Ω), normalized to the surface area of the permeable support (1.12 cm2), and expressed as Ω·cm2.
Bidirectional transport
Bidirectional transport studies were conducted using IPEC-J2 MDR1 cell monolayers cultured on semipermeable Transwell® polyester inserts (Corning Inc., NY, USA, 3460, 1.12 cm2, pore size 0.4 µm). The cells were washed twice with 37°C hHBSS. Before initiation of the experiment, the cells were equilibrated in 37°C hHBSS with or without zosuquidar (ZSQ) (2 µM) for 15 minutes (37°C, 90 rpm). After equilibration, eletriptan HBr (50 µM) was spiked into the donor compartment. Samples were taken from the receiver compartment after 15, 30, 45, 60, 90, and 120 minutes. Samples of 100 uL were withdrawn from the basolateral compartment, while samples of 50 uL were withdrawn from the apical compartment. The withdrawn sample volume was immediately replaced with an equal volume of 37°C hHBSS with or without ZSQ. Donor samples were sampled at the end of the experiment.
The accumulated amount of drug (Q) was calculated for each time-point using Eq. 3.
$$Q={V}_{s}\left(\sum _{n-1}^{n}{C}_{n-1}\right)+{C}_{n}{V}_{t}$$
3
where Vs represents the sample volume, Vt the total volume of the receiver compartment, Cn is the concentration of the sample n.
Steady state flux (Jsteady state) was calculated as the slope of the linear part of the accumulated amount (Q) in the receiver compartment as function of time from Eq. 4.
$${J}_{ss}=\frac{\varDelta Q}{\varDelta t\bullet A}$$
4
where t represents time and A is the area of the permeable support.
The apparent permeability (Papp) was calculated from Eq. 5.
$${P}_{app}=\frac{{J}_{ss}}{{C}_{donor}}$$
5
where Cdonor represents the initial concentration in the donor compartment.
Efflux ratios (ER) were calculated as the ratio of the apparent permeability in the basolateral to apical direction Papp(B-A) and the permeability in the apical to basolateral direction Papp(A-B) using Eq. 6.
$$\text{E}\text{R}=\frac{{\text{P}}_{\text{a}\text{p}\text{p}}\left(\text{B}-\text{A}\right)}{{\text{P}}_{\text{a}\text{p}\text{p}}(\text{A}-\text{B})}$$
6
Sample preparation and quantification of radiolabeled compounds from in vitro experiments
The cells were permeabilized using 0.1% (v/v) Triton X-100 in ultrapure water (ELGA, Buckinghamshire, England) for 10 minutes at room temperature. Thereafter, the cells were scraped off the bottom of the wells, transferred to scintillation vials, and mixed with 2 mL of Ultima Gold 241 TM (Perkin Elmer, MA, USA). The samples were analyzed using a Tri-Carb 2910 TR Liquid Scintillation Analyzer (Perkin Elmer, MA, USA).
Sample preparation and quantification of unlabeled compounds from in vitro experiments
The cells were permeabilized using 50% (v/v) acetonitrile (HPLC LC-MS grade, VWR International S.A.S, Soeborg, Denmark) in ultrapure water (ELGA, Buckinghamshire, England) for 10 minutes at room temperature. Thereafter, the cells were scraped off the bottom of the wells and transferred to HPLC vials. Samples from bidirectional transport experiments were transferred directly into HPLC vials. All samples were stored at -18°C for later analysis. Calibration curves with standards in the range of 1 or 5 to 1000 ng/mL were dissolved in 50% (v/v) acetonitrile in ultrapure water (ELGA, Buckinghamshire, England), or in hHBSS(-) for analysis of bidirectional transport samples.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) were performed using electrospray ionization in positive mode on a micromass Quattro micro™ API tandem quadrupole mass spectrometer (Waters, MA, USA) or an Ultivo triple quadrupole mass spectrometer (Agilent Technologies, CA, USA) coupled to Agilent HPLC system 1100 Series or 1260 Series (Agilent Technologies, CA, USA California), respectively. Applied columns were an InfinityLab Poroshell 120 EC-C18 (3.0 x 50 mm; 2.7 micron, Agilent Technologies, CA, USA) or a Kinetex 2.6 µm XB-C18 (100 Å, 100 x 4.6 mm, Phenomenex Inc., CA, USA). An overview of applied HPLC and MS/MS parameters can be found in Additional file 2 and Additional file 3, respectively. The data acquisition software was MassLynx (V4.1, Waters, MA, USA).
Quantification of total protein content
The protein content of hCMEC/D3 cells was determined for normalization purposes. Cells were washed with ice-cold phosphate buffered saline following cell lysis in cell extraction buffer (FNN0011, Thermo Fisher Scientific, MA, USA) supplemented with 1x cOmplete™ Protease Inhibitor Cocktail (04693116001, Roche, Basel, Schweiz), 1 mM phenylmethylsulfonyl fluoride, and 1 mg/mL pepstatin for 30 minutes on ice. The protein concentration was determined using the bicinchoninic acid (BCA) assay kit (bicinchoninic acid and copper (II) sulphate solution) as directed by the manufacturer. Samples were measured using a SPECTROstar Nano microplate reader (BMG LABTECH, Ortenberg, Germany).
Animals
Male Abcb1a/Abcb1b-eKO1 (P-gp knockout) mice and FVB wild-type counterparts (18–24 g at arrival) were obtained from Shanghai Biomodel Organism Science & Technology Development Co. Ltd (Shanghai, China). Male C57 mice (C57BL/6N) (18–22 g at arrival) were obtained from Beijing Vitalstar Biotechnology Co., Ltd (Beijing, China). Animals were housed in pairs in a temperature-controlled environment (20–24°C) with lighting maintained under a 12-hour light-dark cycle. Animals were habituated for at least seven days prior to surgery with free access to food and water. The experiments were carried out in accordance with the Danish legislation regulating animal experiments; Law and Order on Animal experiments; Act No. 1107 of 01/07/2022 and Act No. 1108 of 01/07/2022 and with the specific license for this experiment issued by the National Authority.
In vivo brain distribution by neuropharmacokinetic assessment and equilibrium dialysis
In vivo experiments were conducted to assess the extent of the BBB transport of eletriptan and to elucidate the involvement of active transport processes. All mice were cannulated in the jugular vein for intravenous drug administration. Following surgery, the animals were allowed to recover for 3 days before administration of test compounds. Eletriptan HBr was dosed in P-gp knockout and wild type mice using a loading dose of 2 mg/kg (as bolus) followed by a 2-hour constant rate infusion of 3 mg/kg (1.5 mg/kg/h) dissolved in 0.9% NaCl. Diphenhydramine hydrochloride was dosed in C57 mice using a 2-hour constant rate infusion of 5 mg/kg (2.5 mg/kg/h) dissolved in 10% HP-β-CD in water. Throughout all studies a dose volume of 10 mL/kg was applied. Serial blood samples were taken from the saphenous vein during the infusion to verify steady state and at the end of infusion. Terminal blood samples were collected from all animals. Brain samples were prepared by homogenization using ultrasonication as described previously (Eneberg et al., 2022) and analyzed together with the plasma samples using LC-MS/MS (40). The lower limit of quantification was determined to 1.0 ng/mL in plasma and 5 ng/g in brain tissue for both eletriptan and diphenhydramine.
Plasma protein and brain tissue binding of eletriptan and diphenhydramine were determined in vitro by equilibrium dialysis using donor test compound solutions of 1 µM incubated in triplicate as described previously (Langthaler et al., 2023) (41). C57 mice were used to prepare blank plasma and brain matrices throughout all binding studies.
The total brain and plasma concentration partition coefficient (Kp) was calculated from Eq. 7.
$${\text{K}}_{\text{p}}=\frac{{\text{C}}_{\text{t}\text{o}\text{t},\text{b}\text{r}\text{a}\text{i}\text{n},\text{s}\text{s}} }{{\text{C}}_{\text{t}\text{o}\text{t},\text{p}\text{l}\text{a}\text{s}\text{m}\text{a},\text{s}\text{s}}}$$
7
where Ctot,brain,ss and Ctot,plasma,ss is the total drug concentration at steady state in brain and plasma, respectively.
The unbound brain and plasma partition coefficient (Kp,uu) was derived from dividing the unbound brain concentration with the unbound plasma concentration at the end of infusion for each animal. The Kp,uu was calculated from Eq. 8.
$${\text{K}}_{\text{p},\text{u}\text{u}}=\frac{{\text{C}}_{\text{t}\text{o}\text{t}, \text{b}\text{r}\text{a}\text{i}\text{n},\text{s}\text{s}} \bullet {\text{f}}_{\text{u},\text{b}\text{r}\text{a}\text{i}\text{n}}}{{\text{C}}_{\text{t}\text{o}\text{t},\text{p}\text{l}\text{a}\text{s}\text{m}\text{a},\text{s}\text{s}} \bullet {\text{f}}_{\text{u},\text{p}\text{l}\text{a}\text{s}\text{m}\text{a}}}$$
8
where fu,brain and fu,plasma is the fraction of unbound drug in the brain and plasma, respectively.
Statistical analysis
Statistical analysis was conducted using GraphPad Prism version 9.4.0 (La Jolla, California, USA). Data are presented as mean ± standard deviation (SD) unless otherwise stated. In vitro experiments were performed in three biological replicates and three technical replicates, unless otherwise stated, where n denotes the number of biological replicates and Ntotal denotes the total number of technical replicates. In vivo experiments were performed in groups of three animals. Statistical analyses were performed by comparing the group means with either two-tailed unpaired Students t-test for comparing two means or by one-way analysis of variance (ANOVA) followed by a Tukey’s multiple comparison test for comparison of more than two means. P-values < 0.05 were considered statistically significant.
{kind=link}