In this section, the surface morphology of electrodes and the interaction of their surfaces with water was investigated. Then, the performance of the electrodes for the H2O2 reaction was studied. Finally, the effect of SDS on the electrode performance was examined.
Figure 1 shows SEM images of the surface of pristine (a) and hydrophobized (b) carbon felts, as well as carbon felts with BPL-PTFE (c) and CBA-PTFE (d) coatings. The untreated felt in (a) consists of randomly oriented carbon fibers with diameters in the range of 5 - 10 mm. The formations of solid PTFE throughout the fiber structure as can be seen in the image of the hydrophobized felt (b).
Similar formations of PTFE, which form a partial film with pores having several micrometers of diameter, can also be found on the surface of the BPL-PTFE electrode (c). This is notably different from the CBA-PTFE substrate (d), where a more uniform porous surface structure can be observed. The contrast in the surface structure can be explained by the difference in bulk density of these two types of CB; that is, 106 g/L for CBA and 464 g/L for BPL as reported by the manufacturers. The same mass load of CBA forms a much thicker coating with a higher porosity in comparison with BPL. Such a thick and porous layer can better absorb PTFE, which is then more uniformly distributed within the bulk of the carbon layer. Unlike CBA, the denser BPL provides a less porous structure to absorb PTFE, which results in partial film formation on the surface of the layer.
Figure 2 shows the image of three electrodes, after 10 cycles from 0.00 to 1.23 V vs. RHE, placed under a layer of water: a pristine carbon felt (a), a carbon felt which was hydrophobized by impregnating with PTFE (b), and a hydrophobized carbon felt coated with a BPL-PTFE layer (c). Cycling the potential of a pristine carbon felt increased its hydrophilicity. When immersed in water, the surface features a uniform appearance with a deep black colour and without attached gas bubbles. In contrast, the carbon felt impregnated with PTFE (b), as well as all CB-PTFE substrate (c), remained their hydrophobicity after cycling which is demonstrated by the layer of gas bubbles when immersed into water, as can be clearly seen in Figure 2 locations (b,c) at the vertical surfaces of the substrates.
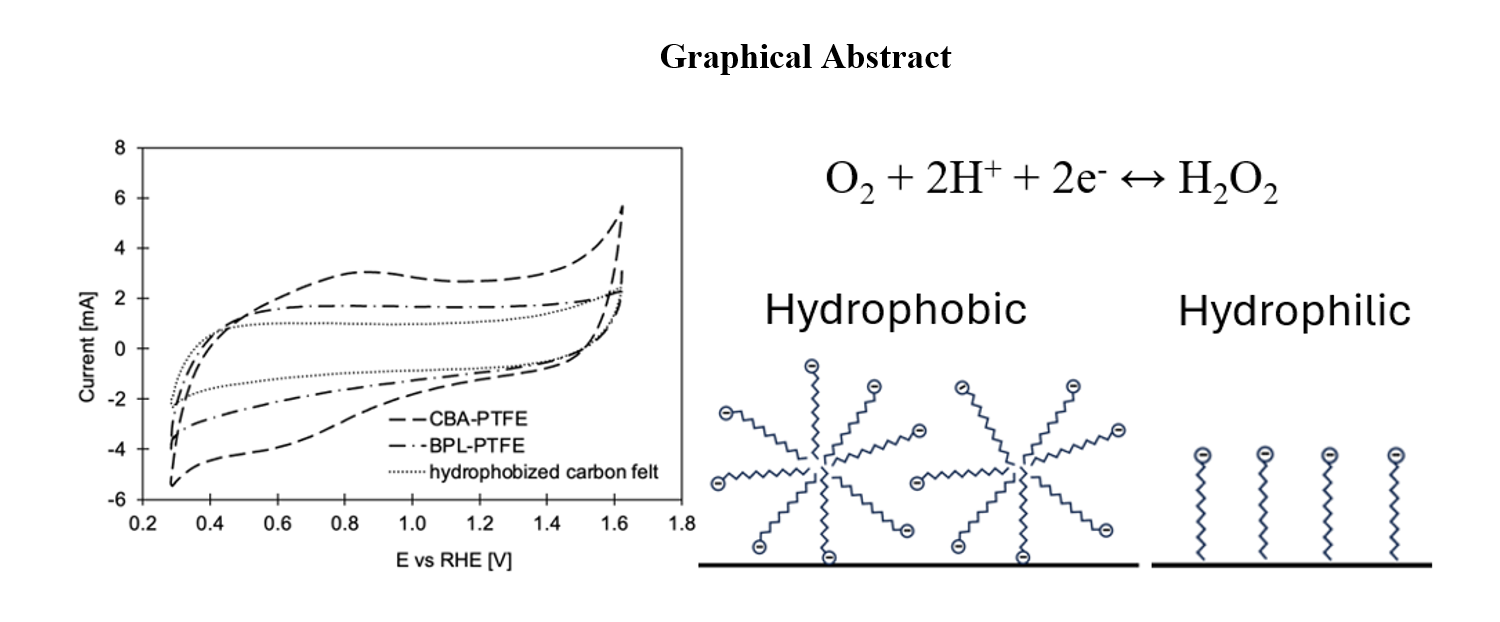
Cyclic voltammograms for carbon felt (pristine and hydrophobized) and for CBA-PTFE and BPL-PTFE electrodes are shown in Figure 3. The voltammograms reveal wide redox peaks between 0.6 V and 0.8 V for the pristine felt and CBA-PTFE electrodes. These peaks are frequently observed on different carbon materials and can be assigned to a quinone–hydroquinone redox reaction of the respective surface functional groups [31]. The peaks disappeared and the double layer charging (capacitive) currents were decreased when the carbon felt was hydrophobized with PTFE as shown in Figure 3a, suggesting a reduction in the solid/solution interface area. In contrast, the currents increased when the PTFE impregnated felt was coated with CB material as can be seen in Figure 3b. The current increase was more apparent for the electrodes with CBA, which also was accompanied with the re-appearance of quinone–hydroquinone peaks. This suggests that CBA possesses respective surface groups and that a larger area of carbon was in contact with the solution. The differences between CBA and BPL in the capacitive behaviour correlates with the bulk density of these materials. It is more than 4 times higher for BPL as discussed earlier. The difference in bulk density suggests that the specific surface is much larger for CBA than for BPL. Since the loading of CB was the same for both types of electrodes, we can anticipate that the carbon area that is available for electrochemical reactions is much larger in CBA-PTFE than in BPL-PTFE electrodes.
Figure 4 shows Nyquist plots for the BPL-PTFE and CBA-PTFE electrodes. The plots reveal a linear region at frequencies below about 1 kHz and a semicircle in the 1-50 kHz range. The observed high frequency resistance was around 3 Ω for both electrodes indicating good electronic conductivity of both substrates.
Figure 5a shows the change in H2O2 concentration during electrolysis for pristine carbon felt, BPL-PTFE and CBA-PTFE cathodes. All three types of electrodes showed activity for the reaction, and the concentration of H2O2 increased with time. The pristine carbon felt had the production rate of H2O2, resulting in concentration a concentration of less than 10 ppm after 40 minutes of electrolysis. The Faradaic efficiency over the first 10 min for this electrode was 27 ± 6%.
Both types of CB electrodes showed an improved H2O2 production rate and higher FE than the pristine carbon felt electrode as can also be seen in Figure 5a. Despite having a lower surface area, the BPL-PTFE electrode produces H2O2 at a faster rate than the CBA-PTFE electrode. After 40 minutes of electrolysis, the concentration of H2O2 was 1.5 – 2 times larger when BPL was used as compared to the electrodes made with CBA. The initial FE of the BPL-PTFE cathodes was about 95%, while for the CBA-PTFE it was only around 50%. However, the FE dropped more significantly over time for the BPL-PTFE electrode, but it still produced more H2O2 than the other electrodes.
The higher activity of the BPL-PTFE electrodes for the H2O2 reaction is also demonstrated by the electrode potentials. Figure 5b shows the cathode potentials that were measured during the experiments reported in Figure 5a. The figure shows that the potential changed somewhat from its initial value of around 190 mV over the first 10-20 minutes, after which it reached a plateau located around 220 mV. There is a significant difference of about 600 mV between the potentials of the BPL-PTFE and the CBA-PTFE electrode. One factor that can be responsible for such a contrast is that the ohmic resistance can be significantly different between these two electrodes. A larger resistance in case of CBA-PTFE would lead to a much higher IR component of the measured potential. However, the high frequency resistance (HFR) was about the same for both electrodes as seen from the EIS measurements in Figure 4. Furthermore, the HFR value of around 3 Ω should only result in 30 mV of ohmic drop for a 10 mA cathodic current, which is an order of magnitude less than the 600 mV difference between the BPL-PTFE and CBA-PTFE electrode potentials. Therefore, we conclude that a higher catalytic activity of the BPL towards the ORR results in lower overvoltage, which is a plausible explanation for the observed potential difference.
The overvoltage can be estimated as a difference between IR-corrected electrode potential under the applied current (about 30 mV more positive than the potentials reported in Figure 5b) and the equilibrium potential that can be calculated from the concentration of hydrogen peroxide using Nernst equation. The concentration of H2O2, achieved after 40 min of electrolysis with BPL-PTFE and CBA-PTFE electrodes, was in the range 10-30 ppm (or mg/L) as seen in Figure 5a, which is on the order of 10-4 M assuming a molar mass of 34 g/mol. According to Nernst equation, the equilibrium potential for the 2-electron ORR is then +0.81 V vs RHE while the standard potential is +0.695 V vs RHE [4]. This implies that the reaction overvoltage of the BPL-PTFE electrodes was about 550 mV at the end of electrolysis. The electrode potential remained at positive values (vs. RHE); i.e., above the potentials of the hydrogen evolution reaction, which explains the high FE observed on this type of electrodes. It is noteworthy that the BPL-PTFE electrodes potentials and FE observed in this work are in agreement with those reported by Karatas et al. [7]. The authors reported about 95% FE of a CB-PTFE electrode in a neutral solution at a potential of 0.25 V vs. RHE (-0.4 V vs. SCE at pH = 7), which is in good agreement with our data reported in Figures 5a and 5b respectively.
The electrode potentials of the BPL-PTFE electrodes are remarkably different to that of the CBA-PTFE electrodes. The electrode potentials were well in the negative values (cf. Figure 5b) suggesting more than 1000 mV overvoltage. These electrode potentials and the respective overvoltages explain the lower FE values for the CBA-PTFE electrodes. At negative potentials of the RHE scale, the ORR competes with the hydrogen evolution reaction and a fraction of the electrical current is spent on producing hydrogen. This is also the case for the pristine carbon felt. We observe that the potential and the FE is even lower than that of the CBA-PTFE electrode, cf. Figures 5a and 5b.
Figure 6 shows the changes in H2O2 concentration during a stability test with both BPL-PTFE and CBA-PTFE electrodes. Here, the electrodes were tested for a longer electrolysis time of 160 min and the solution was replaced every 40 minutes. It can be seen that the difference in the activity of these two types of electrodes remains unchanged. That is, the BPL-PTFE electrode shows higher activity than the CBA-PTFE electrode and no apparent degradation was observed over this operation duration. Considering that the BPL-PTFE electrodes are more active in generating H2O2, we continued our studies with testing the influence of surfactant on the ORR with this electrode.
Figure 7 depicts the effect of SDS on the H2O2 production using BPL-PTFE electrodes and, for the sake of comparison, the pristine carbon felt. In case of the pristine carbon felt, the production of H2O2 was drastically reduced and a very low amount of H2O2, if any, was generated when SDS was added to the solution. Such a strong “poisoning” effect was also observed for a reticulated vitreous carbon cathode when the electrolyte solution contained SDS concentrations in the mM range [22]. This effect was explained by strong adsorption of anionic surfactants resulting in the loss of reaction centers.
In the case of the BPL-PTFE electrodes, the H2O2 concentrations are just moderately lower in the presence of SDS than in the surfactant-free solution, which is remarkably different from the performance of the pristine carbon felt electrode. The initial FE value decreased from 95% to 70% for the electrolyte without SDS while the FE is constantly around 70% in presence of SDS. Hence, the BPL-PTFE electrodes has a relatively better degradation performance in present of the surfactant. Considering the apparent difference in hydrophobicity between the BPL-PTFE electrodes and pristine carbon felt, we hypothesize that this surface property is responsible for the tolerance of PTFE-containing electrodes towards the poisoning effect of SDS on the two-electron ORR.
In order to verify this hypothesis, we compared the H2O2 generating activity of pristine carbon felt and PTFE impregnated (hydrophobized) carbon felt electrodes. Figure 8 shows H2O2 concentration over time for the pristine and the hydrophobized carbon felt electrodes with and without SDS in the solution. The figure reveals that the hydrophobized felt produces considerable amounts of H2O2 even in the presence of SDS. The difference at the end of the reaction time is around 25%. This is notably different from the pristine (hydrophilic) carbon felt which generally produced lower H2O2 concentrations. In the presence of SDS, the hydrophilic carbon felt hardly produces H2O2. The differences in SDS tolerance between the hydrophobic and hydrophilic carbon felt electrodes supports our hypothesis. Hydrophobicity of the surface is responsible for the electrode tolerance towards the poisoning effect of SDS on the two-electron ORR.
The observed difference in ORR Faradaic efficiency on hydrophobic and hydrophilic surfaces can be explained by the different structure of the adsorption layers that surfactants form. Kronberg et al. [32] suggested that adsorption of ionic surfactants on hydrophilic and hydrophobic surfaces have different driving forces and result in remarkably different adsorption layers.
Adsorption of SDS molecules on hydrophobic and hydrophilic surfaces is schematically shown in Figure 9. In case of hydrophobic surfaces, the adsorption process is driven by the tendency of hydrocarbon chains in the surfactant molecules to escape the aqueous environment. With such driving force, the mechanism of surfactant layer formation on the hydrophobic surface is similar to the one for a gas-liquid interface. That is, the formation of a monolayer of surfactant species with tails pointing away from the liquid phase (towards hydrophobic surface in case of solid-liquid interface) as illustrated in Figure 9a. Kronberg et al. [32] noticed that in case of SDS the packing density of the surfactant species in such layers is only about 1/2 of the theoretical close packing density of carbon chains, which suggests that the adsorption layers are not densely packed and are permeable for relatively small molecules like oxygen.
In case of a hydrophilic surface, it does not provide an entity where hydrocarbon chains can escape the aqueous environment. Therefore, arranging the molecules into a monolayer with hydrocarbon chain pointing towards the surface is no longer thermodynamically favourable. Instead, the surfactants form surface-induced self-assemblies. That is, aggregates of entangled molecules with tails pointing towards the internal space of the assemblies and heads towards the hydrophilic surface and the surrounding aqueous solution as depicted in Figure 9b. Such surface aggregates form a much thicker monolayer on the hydrophilic surface which can block the access of oxygen molecules. A comparable mechanism was also proposed to be responsible for the sharp decrease in the ORR activity of reticulated vitreous carbon electrodes in the presence of anionic surfactants like SDS [22]. However, the results of our work suggest that by inducing hydrophobicity on the cathode surface, the adverse effect of surfactants can be mitigated and high Faradaic efficiencies for the H2O2 generation can be achieved.
{kind=link}