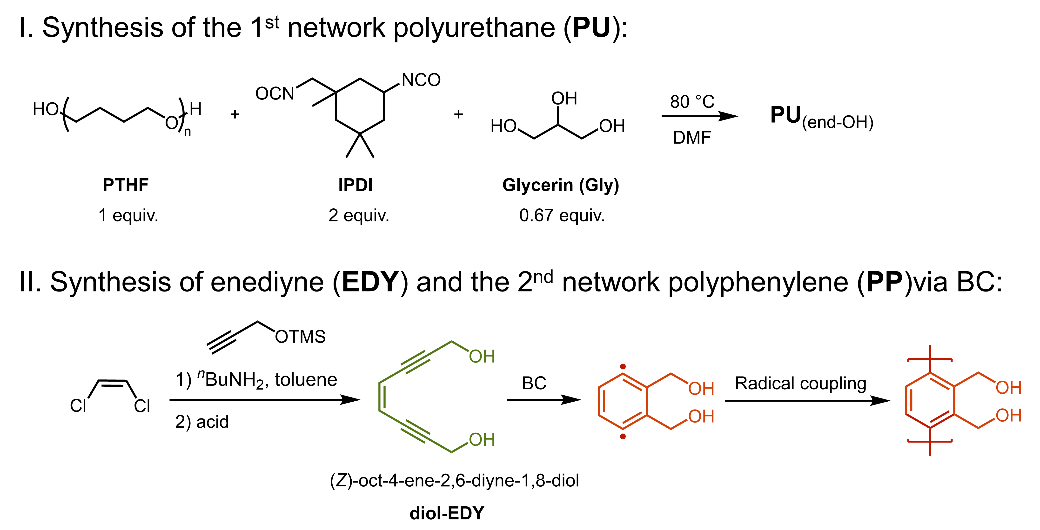
As depicted in Scheme 1, we initiated our studies by synthesizing a polyurethane (PU) network as the basis for the first network, allowing to tune its rigidity and composition. A mixture of polytetrahydrofuran (PTHF, 650 Da), isophorone diisocyanate (IPDI), and glycerol (Gly) (mole ratio of PTHF/IPDI/Gly:1/2/0.67) were heated to obtain the crosslinked PU with dimethyl formamide as the solvent.47 Subsequently, the reaction mixture was cast into a mold, followed by sufficient drying under vacuum and heat. In parallel, the precursor enediyne (EDY) of the interpenetrating network was prepared, representing the basis for the second network. (Z)-oct-4-ene-2,6-diyne-1,8-diol (diol-EDY),48 up to now only scarcely used for the BC process,49–54 was selected, deemed useful due to its stability at ambient conditions, combined with its ease of preparation and the triggerable activation of the BC. For synthesis, the trimethylsilyl (TMS) group was added as a protective moiety on propargyl alcohol, followed by a classical Sonogashira coupling reaction with (Z)-1,2-dichloroethene and subsequent acid-catalyzed deprotection, in turn affording the diol EDY (Figure S1 in supporting information).49 It should be noted that similar to our latest results focusing on diamine EDY ((Z)-octa-4-en-2,6-diyne-1,8-diamine), the final diol-EDY, (Z)-oct-4-ene-2,6-diyne-1,8-diol, is stable in solution with the required concentration owing to its primary alcohol groups inducing intramolecular hydrogen bonding, thus allowing the integration into the first, PU-network without primordial crosslinking reactions.
With the original PU material and the precursor enediyne (diol-EDY) of the second network in hand, the preparation of various samples was conducted as shown in Fig. 2A. After casting and drying mentioned above, a transparent and colorless film was obtained. To remove possibly unreacted monomers, the film was immersed in tetrahydrofuran for 24 h, whereafter all adsorbed initial components for the PU were removed. The so purified PU material was then soaked in the solution of the diol-EDY with ethyl acetate as the solvent (c = 50 mmol/L). With a swelling equilibrium of 48 h, diol-EDY was swollen into the network of PU as expected, together with the solvent ethyl acetate without reaction with the PU-endgroups (targeted as OH), thus allowing a homogeneous distribution of the diol-EDY inside the PU-network. After the swelling film was removed from the solution and dried, the sample PU + EDY was acquired and ready for characterization. The gained weight of PU + EDY compared with the original PU sample was attributed to the gained diol-EDY and showed its content as 4 wt% through calculation. After a thermal-induced BC (150°C for 5 min), the sample IPN (PU + PP) was obtained, wherein the formed 2,5-diradicals form the second networks via radical crosslinking.
To verify the Bergman-cyclization-chemistry and the crosslinking to form the second network, various analyses were conducted. First, the verification of the existence of EDY in the network was probed with electron paramagnetic resonance (EPR) spectroscopy as illustrated in Fig. 2B. Thermally-induced formation of the radicals was demonstrated by the increasing intensity of the carbon-centered radical signals during gradually heating the sample in-situ from 25°C to 175°C in steps of 25°C, revealing the gradual initiation of BC and generation of radical moieties. The double integral (DI) calculated from the EPR spectra at each temperature (25°C, 75°C, 125°C, 175°C) is directly correlated to the radical concentration in the sample. The DI was plotted against the temperature, which is shown in the inset of Fig. 2B, clearly proving the formation of radicals inside the IPN by thermal initiation of the embedded EDYs.
As BC is an exothermic reaction, differential scanning calorimetry (DSC) was measured with the samples PU + EDY and IPN (PU + PP) to probe the progress of the BC, indicated by a decreasing exothermic profile as the reaction proceeded. Both samples were heated to 250°C, followed by cooling to 25°C under an atmosphere of N2. As shown in Fig. 2C, for the intermediate sample PU + EDY (black line), the exothermic peak (~ 140°C) was attributed to a thermally triggered BC of the enediyne moiety. For the informed interpenetrating sample IPN (PU + PP), the flat curve (red line) during heating further proved the completion of BC of the gained enediyne inside. Furthermore, the occurrence of a BC was proven by IR-spectroscopy, indicating the chemical changes occurring during BC, changing from the EDY-moieties to the poly(aromatic) polymers formed after BC (see Fig. 2D/E). The comparison of the spectra between sample PU + EDY (black line) and the IPN (PU + PP) (red line) showed a decreased stretching band of the ene (C ≡ C) at 2271 cm− 1 after the formation of IPN, giving solid evidence about the consumption of the alkynyl-groups after BC. Besides, a characteristic absorption at around 810 cm− 1 in the sample IPN could be attributed to the CH bending of the conjugated benzene ring system. It is worth mentioning that, compared to the CH bending on the 1,2-substituted benzene, the absorption presented here at a higher wavenumber (810 cm− 1) revealed the formation of the 1,2,3,4-substituted benzene moieties as a result of free radical polymerization in the presence of diradical intermediates. In addition, as shown in Table 1, swelling experiments proved the formation of a second network by BC: the presence of the interpenetrating composition decreased the swelling degree of the network, from 131% for PU to 119% IPN (PU + PP). Therefore, the existence of EDY which was swollen into the PU network, together with the informed skeleton of IPN (PU + PP) via BC, was verified.
With IPN (PU + PP) in hand, the difference in the mechanical properties compared to the original PU was examined, as the formation of a second network inside the first PU-network was expected to yield different mechanical properties. To avoid eventual errors caused by the soaking process of sample IPN (PU + PP) in EDY/ ethyl acetate solution, PU characterized in this paper was treated precisely with the same soaking process in a blank solvent (ethyl acetate) without EDY. As illustrated in Fig. 3, the mechanical properties of both samples were studied by stress − strain characterization and cyclic stress − strain investigations. Their determined strain at break, stress at break, and dissipated energy values at each strain are depicted in Table 1. Correspondingly, rectangular specimens with a length of 40 mm, a width of 4 mm, and a thickness between 0.2 and 0.4 mm were prepared. Three specimens for each sample were measured on average with a uniform stretching speed of 20 mm min− 1. For the two elastomer samples, an increase of the tensile stress was observed from 13.9 MPa for PU to 15.0 MPa for IPN (PU + PP), while the extensibility slightly decreased from 679–672%. The slightly improved mechanical properties could be attributed to the existence of the second network polyphenylene, inducing structural interpenetration and physical entanglement, thus adding its rigidity.55,56 In the research of IPNs, higher tensile strength and lower elongation are common.57 Besides, cyclic stress − strain investigations of both samples were performed at room temperature to study the energy dissipation capacity related to the bonding interactions. Therefore, cyclic loading and unloading at a maximum strain of 50%, 100%, 150%, and 200%, being far below the strain at break, were performed continuously without rest. The area between the load and unload stress-strain curves equals the dissipated energy. Compared with PU, the integral area of the corresponding hysteresis loop of IPN (PU + PP) was increased about 48 times on average, further indicating the impact of the coexisting networks on the mechanical properties in its now interpenetrating form.
Table 1
Overview of dissipated energy of sample PU and IPN (PU + PP) at a maximum strain of 50%, 100%, 150%, and 200%, together with strain at break, stress at break of each sample, and swelling degree of two samples in toluene.
|
Sample
|
Dissipated energy (MJ/m3)
|
Fracture strain (%)
|
Fracture stress (MPa)
|
Swelling (%)
(toluene)
|
|
Strain 50%
|
Strain 100%
|
Strain 150%
|
Strain 200%
|
|
PU
|
16.3
|
35.0
|
55.7
|
80.0
|
679
|
13.9
|
131
|
|
IPN (PU + PP)
|
27.6
|
53.4
|
77.9
|
102.0
|
672
|
15.0
|
119
|
As illustrated in Fig. 4, thermogravimetric analyses (TGA ) was applied further to study the thermal stability of the original PU and the obtained IPN (PU + PP) through the analysis of the mass change. Their determined temperature and mass loss values in different stages of decomposition, together with the residue percentages at 600°C are depicted in Table 2. The decomposition processes of both samples were quite comparable. The 5% decomposition of PU (T5wt%) happened at around 281°C, but for IPN (PU + PP), the temperature needed for the same 5% decomposition was increased to 286°C. The improved thermostability could be related to the interpenetrating network composed of PU and PP which is a supramolecular structure at the micro-level, retarding the decomposition process. Additionally, two stages of degradation were recorded for both samples with similar maximum degradation temperatures (for Tmax1: 338°C, for Tmax2: 397°C), but with respectively different proportions of mass loss in each of the two stages. During the first stage, focusing on the dissociation of urethane linkages for both samples, IPN displays a lower mass loss compared to PU. A more drastic decrease in mass loss and a lower maximum weight loss rate for IPN were detected during the second stage involving the rupture of ester linkages and fatty acid chains of the polyurethane soft segments.58,59 Therefore, TGA and the corresponding DTG verified the improved thermostability owing to the existence of PP polymer interpenetrated with PU polymer. The compact network formed by interpenetration and physical entanglement of the two components makes the bond rupture relatively more difficult. Increased residue at 600°C for IPN stemmed from the carbon material polyphenylene.
Table 2
Thermal properties of PU and the IPN (PU + PP).
|
Sample
|
T5wt%/°C
|
T15wt% /°C
|
1st decomposition
|
2nd decomposition
|
Residue (600 °C) /wt%
|
|
Mass loss/ wt%
|
Tmax /°C
|
Mass loss/ wt%
|
Tmax /°C
|
|
PU
|
281
|
305
|
62.2
|
338
|
35.9
|
397
|
0.1
|
|
IPN (PU + PP)
|
286
|
309
|
61.9
|
338
|
32.9
|
398
|
3.3
|
To investigate the surface morphologies, atomic force microscopy (AFM) analysis in tapping mode was performed on three thin films throughout the synthetic route: the original PU, intermediate PU + EDY, and final IPN (PU + PP) (Fig. 5). For PU, the surface morphology exhibited as a flat surface with some tiny gullies, but no phase separation, which is the result of thermodynamic compatibility between the hard and soft segments in the PU chains synthesized here. Similarly, an isotropically flat surface without a preferred direction was detected with IPN (PU + PP), revealing its surface morphology in a random arrangement as a uniform mixture. For PU + EDY, segregated domains were observed clearly on the surface, with the texture providing a height of ~ 62 nm. This arises from the insertion of the diol-EDY-monomer and is in good agreement with the designed manufacturing process yielding the final networks as shown in the structural illustration of three samples (right side).
{kind=link}