Colorectal cancer (CRC) is globally recognized as the third most prevalent form of cancer and has the second highest fatality rate among all types of cancer[16]. This research demonstrates that the early detection of CRC can lead to a reduction in mortality rates. Colonoscopy, recognized for its high sensitivity and specificity, is the preferred approach for screening CRC. However, the accessibility of this procedure is influenced by the patient's financial means and the proficiency of the performing physician[17]. Additionally, due to variations in anatomy and histology, left-sided CRC (arising from the splenic flexure, descending, and sigmoid colon) and right-sided CRC (originating in the cecum, ascending colon, and hepatic flexure) are distinct clinical entities with notable disparities in treatment efficacy and prognosis[18–19]. Numerous studies have corroborated that right-sided CRC tends to have a more unfavorable prognosis compared to left-sided CRC[20].
Therefore, accurate and cost-effective early screening for both types of colorectal cancer constitutes a primary objective in ongoing research endeavors. Guo W and his team utilized single-cell sequencing to map the differences between the left and right halves of colorectal cancer for the first time. Digging deep into cell types, transcriptomics, and the tumor microenvironment, the team analyzed the cellular and molecular features of the two diseases and discovered a set of genes that are specifically expressed in left-sided CRC. We learned from the research of his team that the differences in cancer cell signaling pathways, tumor microenvironment and immune cell status play a crucial role in the prognosis of left and right colorectal cancer.[20].
In previous retrospective systematic studies, researchers have generally found that the TP53 mutation rate of left colorectal cancer is pretty high, while the BRAF, PIK3CA, CTNNB1, SMAD4 and KRAS mutation rates of right colorectal cancer are pretty high, and the incidence of mucinous histology, CIMP and MSI are all very high [21–27].
DongXF et al. used immunohistochemistry to explore the expression of WTAP(up-regulated in malignant tumors and considered to be a cancer gene) in the tissues of CC patients, and results showed that WTAP was the highest expression in samples of left colorectal cancer, showing a positive correlation, and a negative correlation with poorly differentiated tumors, providing new ideas for early screening and treatment of left colorectal cancer [27]. Because of the different molecular mechanisms of the two diseases, there are differences in prognosis and treatment. Currently, surgical resection is still a common choice for non-metastatic colorectal cancer, but it has been reported that the incidence of postoperative complications and surgical site infection in the right half of colorectal cancer is relatively low, and there is no significant difference in other postoperative symptoms [28–29]. Overall, right colorectal cancer has a poor prognosis and a higher mortality rate than left colorectal cancer, which may be due to the higher likelihood of positive MSI in right colorectal cancer [30] as well as the fact that left colorectal cancer can be screened relatively early by endoscopy [30].
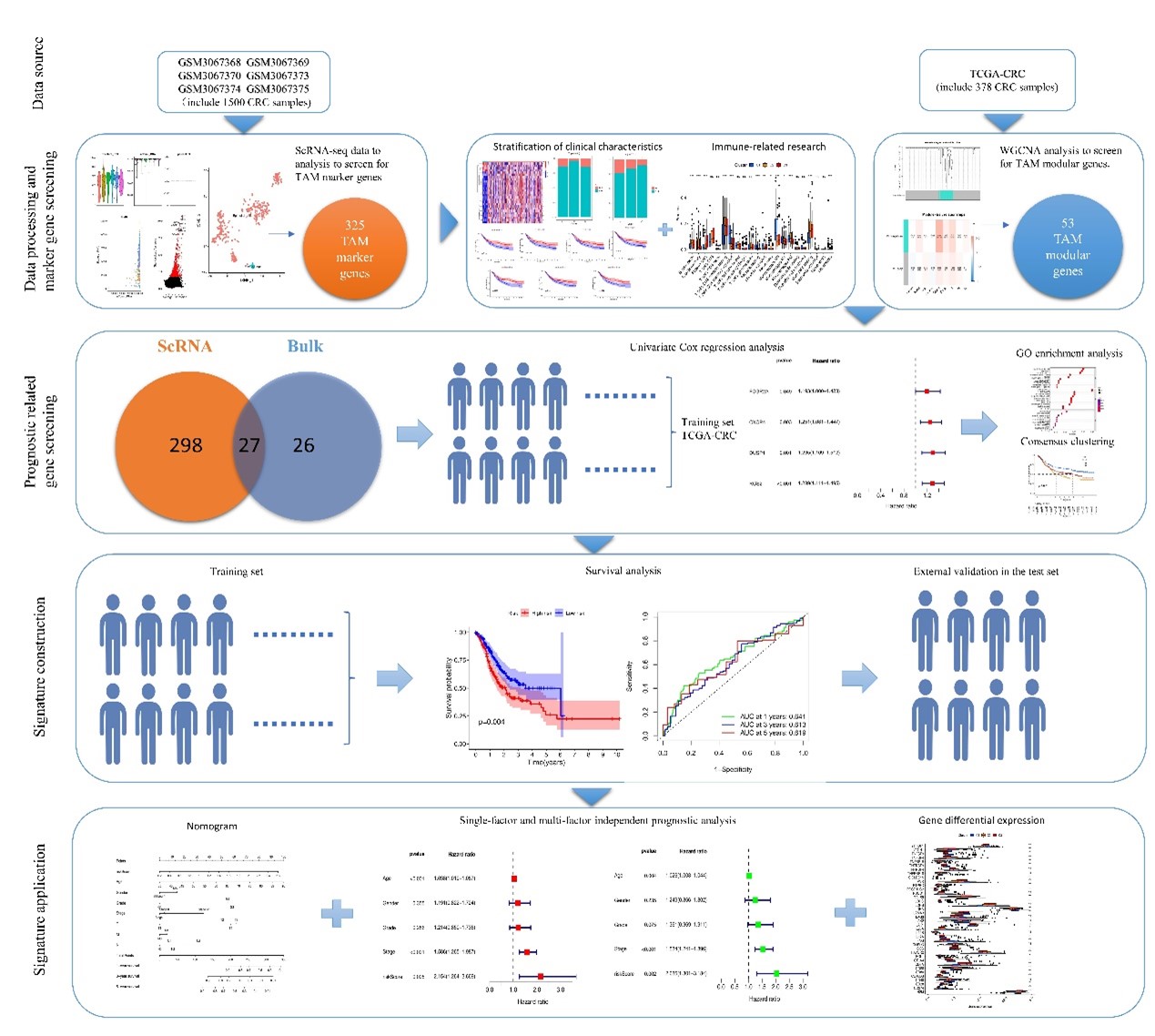
Single-cell sequencing technology is a highly effective and innovative tool to decipher molecular maps based on single cells, which has an increasingly extensive application and promising prospect in cancer research [31]. In our study, we analyzed cellular and genetic differences between LCC and RCC using single- cell sequencing. In addition, we constructed a macrophage-based prognostic model for CC patients based on single-cell sequencing data. WGCNA was used to further screen out four TAM-associated prognostic genes related to CRC prognosis, namely FCGR2A, CXCR4, DUSP1 and RGS2. These genes were utilized to build our prognostic model. Our study also analyzed the infiltration of immune cells, and we found that some differentially expressed genes in patient macrophages were significantly associated with survival. ROC curve showed that the area under AUC at 1, 3 and 5 years were 0.641, 0.613 and 0.619 respectively.
Analyses of age, sex, grade, stage, and risk score were conducted using both univariate and multivariate methods to assess the model's prognostic independence. The findings indicated that the risk score stands out as a significant prognostic element. The calibration curves were utilized to appraise the model's performance, further corroborating its dependability. Prior research, including single-cell RNA sequencing, has illustrated that macrophage diversity increases in inflammatory contexts like cancer[32]. The advancement of CRC is frequently linked to a systemic inflammatory reaction, which involves the release of inflammatory mediators into the bloodstream. These mediators contribute to tumor growth and dissemination via various pathways, such as enhancing tumor cell proliferation and conditioning premetastatic sites to support further metastasis[33–34].
A survival analysis focusing on immune checkpoints was conducted, revealing that certain checkpoints, including YTHDF1, PVR, and LDHA, are overexpressed in the C3 group, suggesting potential novel targets for immunotherapeutic interventions. The expression levels of immune checkpoint genes like CD80, CTLA4, ICOS, IFNG, JAK2, LAG3, and TNFRF9 vary among patients, with increased expression being associated with improved survival outcomes. Notably, CD80, a costimulatory molecule, is recognized as a highly effective factor for enhancing immune cell activity and curbing the advancement of cancer[35].
While our research has yielded novel single-cell insights, it is not without its constraints. The absence of immunohistochemical assays and additional verification for model genes is a notable gap in this study. It is imperative that future studies are multi-centric to substantiate our findings. Moreover, due to restrictions in threshold determination and methodological constraints, a comprehensive analysis of all sequenced genes was not feasible, potentially impacting the effective screening and identification of macrophage-related genes. The sample size used in this study is relatively limited, which may affect the model's accuracy. The AUC for 1, 3, and 5 years was 0.641, 0.613, and 0.619, respectively, suggesting that there is a need to either increase the sample size or utilize multiple testing sets to further validate the model.
{kind=link}