Body weight
The body weights (BWs) of the subjects constantly increased during the intervention. Notably, the hindlimb unloading groups (TSS, Van + TSS, and GSW + TSS) exhibited a lower growth rate than their reference groups (Con, Van, and GSW). In addition, after 35 days feeding, the gap between BWs of rats from group Van + TSS and that from group Van had significantly increased when compared with the gap between BWs of rats from group TSS and from group Con(Fig. 1).
Microgravity exposure induced lipid deposition and inflammation within the liver
The liver is the central metabolic organ and is the first bypass organ through which nutrients are absorbed in the human body and rodents; moreover, the liver is susceptible to and prone to complications from microgravity when stimulated by TSS. As characterized by liver hematoxylin-eosin (H&E) staining, TSS treatment and vancomycin administration resulted in a loose tissue structure and a decreased total cell area, and vancomycin administration aggravated the liver tissue dispersion in rats treated with the tail suspension (Fig. 2a and c). Moreover, liver oil red O staining indicated that TSS treatment promoted lipid deposition in the liver of rats from group TSS, GSW + TSS, and Van + TSS. As expected, vancomycin administration also accentuated liver lipid deposition in the group Van + TSS (Fig. 2b and d). In agreement with the above findings, the levels of total cholesterol (TC) in liver tissue homogenates were significantly greater in group which combined TSS treatment with vancomycin administration (Fig. 2e), and total triglyceride (TG) was significantly greater in all TSS-related treatment groups (including the TSS, GSW + TSS, and Van + TSS groups) (Fig. 2f). To further evaluate the degree of liver damage, the concentrations of serum alanine transaminase (ALT) and aspartate aminotransferase (AST) were measured. Unexpectedly, both the two serum biomarkers did not significantly increase in TSS-related groups (Fig. 2g, 2h). Oxidative-related factors in the liver were subsequently evaluated, including malondialdehyde (MDA), glutathione peroxidase (GSH-Px), and superoxide dismutase (SOD). As shown in Fig. 2i, the levels of liver MDA in group Van, TSS, Van + TSS and GSW + TSS were significantly greater than those in their comparable groups. Moreover, compared with corresponding controls, the concentrations of GSH-Px in liver homogenates from rats of group Van, Van + TSS and GSW + TSS were also obviously greater (Fig. 2j). In contrast, the levels of SOD in liver homogenates from group GSW + TSS were markedly lower than it from group GSW (Fig. 2k). Furthermore, the concentrations of proinflammatory cytokines (TNF-α, IL-6, and IL-1β) in liver homogenates were also detected. The results showed that the levels of TNF-α in group Van were greater than that in group GSW, and were lower in both of group Van + TSS and GSW + TSS than those in their comparable group Van and GSW (Fig. 2l). Moreover, the levels of IL-6 in group TSS, Van + TSS, and GSW + TSS were significantly greater compared with their control groups (Fig. 2m). Simultaneously, the levels of IL-1β exhibited a significant increase in the GSW + TSS group, whereas the other TSS-related groups (TSS and Van + TSS) did not exhibit any changes (Fig. 2n).
Integrality of the colonic mucosa
The colon mucosa is composed of the intestinal epithelium, lamina propria, muscle and so on. As shown in H&E images of colon, the rats subjected to TSS treatment and vancomycin administration owned a colon which presented with sparse and narrow mucosa, irregular crypt bases, rough epithelium surfaces, and damaged lamina propria in some niches whereas their control rats did not (Fig. 3a). In contrast to rats who were dealt with vancomycin administration, rats suffering from TSS treatment had better colon mucosa, who only damaged in partial segment, whereas vancomycin administration damaged the whole mucosa. Moreover, we detected 7 serum/plasma biomarkers associated with the permeability of intestinal barrier function, namely, lipopolysaccharide (LPS), lipopolysaccharide binding protein (LBP), D-lactate (D-Lac), diamine oxidase (DAO), secretory immunoglobulin A (SIGA), calprotectin (CALP) and chromogranin A (CGA) (Fig. 3b). Interestingly, the serum microbial endotoxin LPS was significantly increased in group TSS (p = 0.00014), and the serum D-lactate was increased in group TSS (p = 0.013) and GSW + TSS (p = 0.012). In response to the endotoxin, the concentration of serum LBP was obviously lower in group TSS (p = 0.04),
Van (p = 2.86×10− 4), Van + TSS (p = 0.038) and GSW + TSS (p = 4.98×10− 7). Moreover, the concentrations of serum intracellular enzyme DAO whom oxidizes diamines (histamine, putrescine and cadaverine) were also markedly lower in group TSS (p = 0.0036) and Van + TSS (p = 5.92×10− 4) than their comparable groups. Unexpectedly, the SIGA did not significantly change during TSS or vancomycin treatment, except for a mild decrease in group TSS (p = 0.16). CALP is a calcium-containing protein derived from neutrophils and macrophages which plays a role in anti-inflammatory and immunologic enhancement. Interestingly, the plasma CALP was expressed in TSS-dependent manner. Namely, the levels of plasma CALP were consistently lower in rats among group TSS (p = 4.38×10− 5), Van + TSS (p = 8.37×10− 6) and GSW + TSS (p = 1.04×10− 5). In addition to CALP, another immune related protein-CGA, had increased concentrations in plasma from rats among group TSS (p = 0.024) and GSW + TSS (p = 0.044).
Dysfunction of the gut microbiota caused by microgravity exposure
The relative abundance of fecal microbiota in group TSS was characterized by decreasing proportions of genus Lactobacillus, Prevotella and increasing genus Shigella, Ruminococcus, Desulfovibrio, and Coprococcus when compared with group Con. Reference rats treated with vancomycin can destroy microbiota. Consistent with these findings, when rats in the Van group were treated with vancomycin, the intestinal microbiota exhibited a remarkable increase in the abundance of the detrimental genera Shigella, Enterococcus, Morganella, Desulfovibrio, and Phascolarctobacterium and few comparable beneficial genera Ruminococcus, Oscillospira and Coprococcus compared with those in the GSW group. Furthermore, when the plants were overlaid with microgravity, the Van + TSS group presented a lower proportion of Lactobacillus and greater abundances of Shigella and Enterococcus than did the Van group (Fig. 4a). The taxonomic classification tree evolved with tail-suspension and vancomycin treatment, during which the phylum Gammaproteobacteria evolved into the group Van and Van + TSS, whereas TSS treatment influenced mainly gut microorganisms at the genus level; these changes included the evolution of Lactobacillus, Streptococcus, Coprococcus, Oscillospira, and Shigella (Fig. 4b).
The microbial α diversity measures the richness, diversity, and evenness of taxonomy, of which the Chao1 estimator reflects the microbial richness by calculating “Singleton” together with “Doubleton” in the operational taxonomic unit (OTU). According to the sparse curves, the Chao1 indices increased with sequencing depth and equilibrated at nearly 20,000 depths, which indicated that the obtained OTUs were sufficient for feature extraction. In particular, the Van and Van + TSS groups had markedly lower α diversity indices than did the other groups, and the Con group had the highest α diversity index (Fig. 4c). In contrast to that of α diversity, the microbial β diversity differed according to treatment. Except for the Van and Van + TSS groups, which were strongly different from the control group, the other groups exhibited similar β diversity in terms of microbial community structure (Fig. 4d).
The core and unique OTUs were counted in a Venn diagram. Herein, the TSS group had markedly fewer unique OTUs than did the Con group (from 4076 to 1419). Similarly, the number of unique OTUs in the Van combined with Van + TSS group also decreased compared with that in the GSW or GSW + TSS groups (Fig. 4e). Finally, biomarkers of the microbiota from each group were screened via linear discriminant analysis (LDA) effect size (LEfSe) analysis, which integrates the nonparametric Kruskal‒Wallis test and Wilcoxon rank sum test with linear discriminant analysis (LDA). According to the LDA scores, the intestinal microbial communities of rats in the TSS group were characterized by the genera Streptococcus, Candidatus_Solibacter, Tepidimicrobium, Butyrivibrio, Anaerotruncus and some other unknown genera. Furthermore, with increasing vancomycin concentration, the microbial ecology of rats in the Van + TSS group was enriched for the genera Bilophila, Sutterella, Akkermansia, and Enterococcus and some other unknown genera (Fig. 4f).
Metabolic profiling on liver and colon
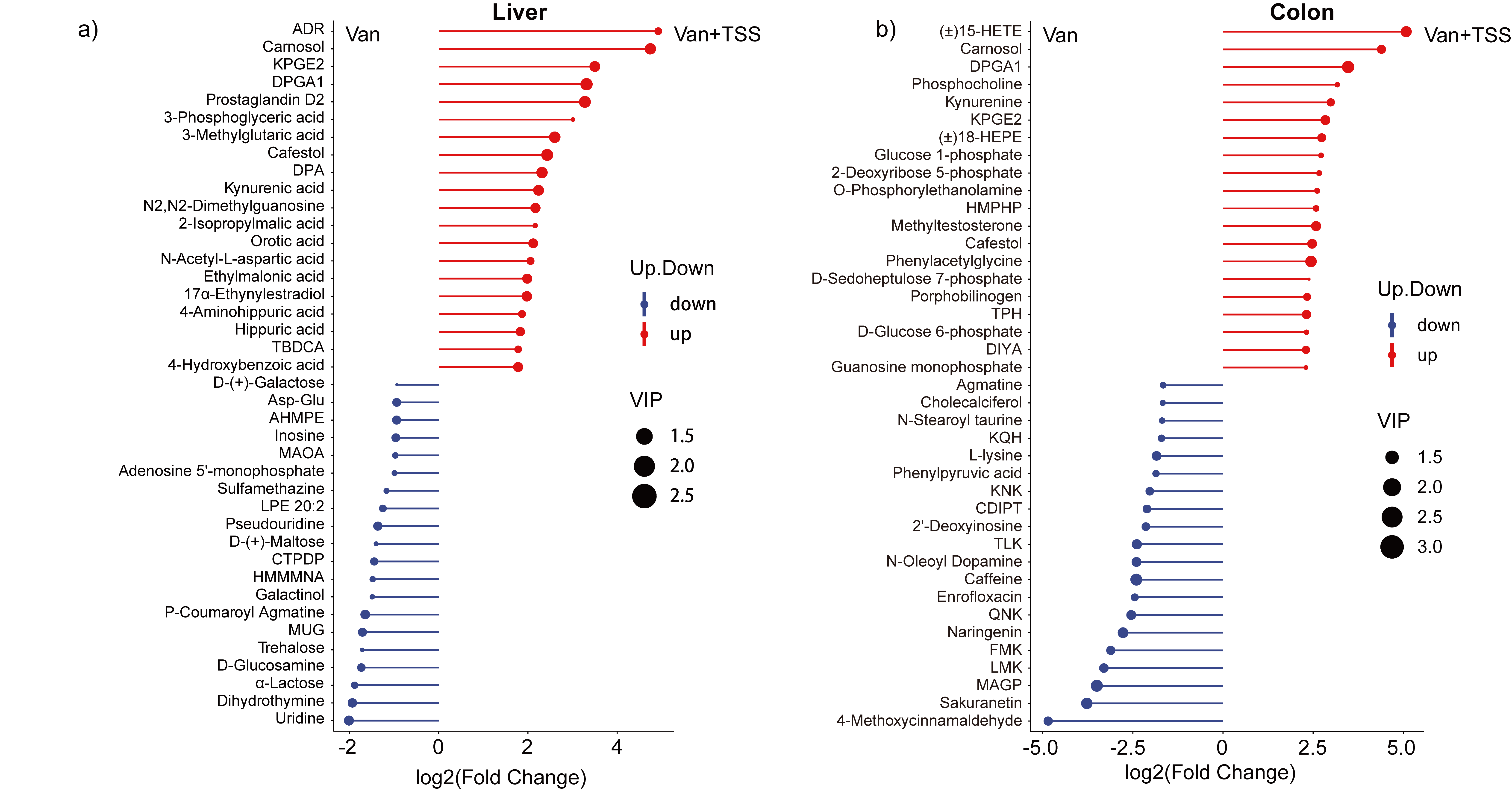
The supernatants extracted from rats’ liver and colon were subjected to metabolic profiling via ultrahigh-performance liquid chromatography coupled with Orbitrap Exploris mass spectrometry (UPLS-MS2) to reveal changes in the gut-liver axes. Notably, the TSS treated groups, including group TSS, GSW + TSS and Van + TSS, exhibited similar projections in hepatic metabolic profiles and processed distinguishable distances far away from their reference groups (Con, GSW, and Van) which distributed along with the x-axis in the score projection space which was modeled by orthogonal signal correction-orthogonal partial least squares-discriminant analysis (O2PLS-DA). Furthermore, vancomycin treatment induced distinct clustering which distributed along with the y-axis for liver metabolism when combined with TSS (Fig. 5a). In contrast to liver metabotyples, colon metabolic profiles had shown to be strongly related to the impactions of TSS and sensitive to vancomycin treatment as characterized by distinct clustering which distributed along with x-axis (Fig. 5e). Liver and colon metabolic shifts were further verified by heatmaps of metabolite intensity in which liver metabolic profiles appeared in according to TSS associated clustering whereas the colon presented vancomycin related clustering (Fig. 5b, f). Furthermore, the characteristic metabolites were screened by the VIPs (predictive variable importance in the projection from O2PLS-DA), FC (fold change), and P value (one-way ANOVA with standard Bonferroni correction) (Table 1). As shown by the set of liver- and colon-specific metabolites, a total of 1265 metabolites (907 from the positive ion mass spectrum and 358 from the negative ion mass spectrum) were identified in the liver and colon, among which 164 metabolites from liver were significantly upregulated and 111 downregulated along with TSS treatment. Yet, 270 upregulated and 74 downregulated liver metabolites were associated with treatment which combined TSS with vancomycin administration. In contrast, few metabolites (a total of 17) from colon shown to be sensitive to TSS treatment, but many (62 upregulated and 99 downregulated in the Van + TSS group versus the Van group) changed with vancomycin treatment which could destroy the microbiota.
Table 1
Overview of significant metabolites between differential treatment groups according to VIP, FC and P value
| Compared Treatments1 | Tissue | Num. of Total Ident.2 | Num. of Total Sig.3 | Num. of Sig.Up4 | Num. of Sig.down5 |
| Positive | Negative | Positive | Negative | Positive | Negative | Positive | Negative |
| Con.vs.GSW | Liver | 907 | 358 | 41 | 31 | 15 | 22 | 26 | 9 |
| Van.vs.GSW | 907 | 358 | 69 | 33 | 40 | 19 | 29 | 14 |
| Van + TSS.vs.Van | 907 | 358 | 211 | 133 | 151 | 119 | 60 | 14 |
| TSS.vs.Con | 907 | 358 | 199 | 75 | 117 | 47 | 82 | 28 |
| GSW + TSS.vs.GSW | 907 | 358 | 319 | 113 | 179 | 90 | 140 | 23 |
| GSW + TSS.vs.Van + TSS | 907 | 358 | 136 | 75 | 44 | 19 | 92 | 56 |
| Con.vs.GSW | Colon | 907 | 358 | 48 | 10 | 23 | 3 | 25 | 7 |
| Van.vs.GSW | 907 | 358 | 241 | 104 | 181 | 74 | 60 | 30 |
| Van + TSS.vs.Van | 907 | 358 | 111 | 50 | 40 | 22 | 71 | 28 |
| TSS.vs.Con | 907 | 358 | 14 | 3 | 6 | 2 | 8 | 1 |
| GSW + TSS.vs.GSW | 907 | 358 | 206 | 62 | 77 | 28 | 129 | 34 |
| GSW + TSS.vs.Van + TSS | 907 | 358 | 246 | 110 | 73 | 30 | 173 | 80 |
Table 1
Overview of significant metabolites between Compared Treatments screening by VIP, FC and P-value
| Compared Treatments1 | Tissue | Num. of Total Ident.2 | Num. of Total Sig.3 | Num. of Sig.Up4 | Num. of Sig.down5 |
| Positive | Negative | Positive | Negative | Positive | Negative | Positive | Negative |
| Con.vs.GSW | Liver | 907 | 358 | 41 | 31 | 15 | 22 | 26 | 9 |
| Van.vs.GSW | 907 | 358 | 69 | 33 | 40 | 19 | 29 | 14 |
| Van + TSS.vs.Van | 907 | 358 | 211 | 133 | 151 | 119 | 60 | 14 |
| TSS.vs.Con | 907 | 358 | 199 | 75 | 117 | 47 | 82 | 28 |
| GSW + TSS.vs.GSW | 907 | 358 | 319 | 113 | 179 | 90 | 140 | 23 |
| GSW + TSS.vs.Van + TSS | 907 | 358 | 136 | 75 | 44 | 19 | 92 | 56 |
| Con.vs.GSW | Colon | 907 | 358 | 48 | 10 | 23 | 3 | 25 | 7 |
| Van.vs.GSW | 907 | 358 | 241 | 104 | 181 | 74 | 60 | 30 |
| Van + TSS.vs.Van | 907 | 358 | 111 | 50 | 40 | 22 | 71 | 28 |
| TSS.vs.Con | 907 | 358 | 14 | 3 | 6 | 2 | 8 | 1 |
| GSW + TSS.vs.GSW | 907 | 358 | 206 | 62 | 77 | 28 | 129 | 34 |
| GSW + TSS.vs.Van + TSS | 907 | 358 | 246 | 110 | 73 | 30 | 173 | 80 |
| The significant metabolites were screened by VIP (Variable Importance in the Projection), FC(Fold Change), and P-value, VIPs were from O2PLS-DA (orthogonal signal correction-orthogonal partial least squares-discriminant analysis);P-value was calculated by one-way ANOVA with standard Bonferroni correction. The threshold values were set as VIP > 1.0, FC > 1.2 or FC < 0.833 and P-value < 0.05. 1. Compared Samples: compared treatments which compare the former treatment with the latter; 2. Num of Total Ident: total numbers of identified metabolites from MS༛3. Num of Total Sig: total numbers of significant metabolites; 4. Num of Sig Up: total numbers of significant up-regulated metabolites; 5. Num of Sig down: total numbers of significant down-regulated metabolites。 |
The significant metabolites were screened by variable importance in projection (VIP), fold change (FC), and P value. VIPs were identified via orthogonal signal correction-orthogonal partial least squares-discriminant analysis (O2PLS-DA). The P value was calculated via one-way ANOVA with standard Bonferroni correction. The threshold values were set as VIP > 1.0, FC > 1.2 or FC < 0.833 and P value < 0.05. 1. Comparison of Samples: Comparisons of the former treatment with the latter; 2. Num of total identity: total number of identified metabolites from MS; 3. Num of total Sig: total number of significant metabolites; 4. Num of Sig Up: total number of significantly upregulated metabolites; 5. Num of Sig: total number of significantly downregulated metabolites.
Metabolite pathway enrichment analysis (MPEA) was performed for physiological interpretation base on metabolites screening from various treatments. The pathways mostly enriched for TSS related metabolites derived from liver were defined as aminoacyl-tRNA biosynthesis; mineral absorption; serotonergic synapse; valine, leucine and isoleucine biosynthesis; tryptophan metabolism; and vitamin B6 metabolism. And, the colon-derived metabolites were associated with the pathway of gastric acid secretion (Table 2). Furthermore, the rats whom received vancomycin administration in combination with TSS treatment enriched pathways which hit many proinflammatory metabolites. Namely, the liver exhibited changes in arachidonic acid metabolism, the oxytocin signaling pathway, and renin secretion, and the colon exhibited changes in glycerophospholipid metabolism (Table S1).
Table S1
KEGG enrichment of characteristic metabolites between the Van + TSS group and the Van group.
| Tissue | MapID | MapTitle | Pvalue | MetaIDs |
| Liver | map00590 | Arachidonic acid metabolism | 0.015 | Prostaglandin E2; Prostaglandin H2; Lipoxin B4; Thromboxane B2; Prostaglandin D2; 16(R)-HETE; Arachidonic acid; Prostaglandin J2; 5-OxoETE |
| map04924 | Renin secretion | 0.044 | Prostaglandin E2; Adenosine; Adenosine 5'-monophosphate |
| Colon | map00564 | Glycerophospholipid metabolism | 0.009 | O-Phosphorylethanolamine; Phosphoethanolamine; Phosphocholine; Cytidine 5'-diphosphocholine |
| MapID: map ID of the enriched KEGG pathways. MapTitle: title of enriched KEGG pathways. P value: Overrepresentation analysis was implemented using a hypergeometric test to evaluate whether a particular metabolite set was represented more than expected by chance within the given compound list. One-tailed p values are provided after adjusting for multiple testing. MetaIDs: List of the input metabolites that participated in the KEGG pathway. |
Table 2
KEGG enrichment of liver metabolites derived from the liver and colon between the TSS and Con treatment groups
| Tissue | MapID | MapTitle | Pvalue | MetaIDs |
| Liver | map00970 | Aminoacyl-tRNA biosynthesis | 0.006 | L-Asparagine; L-Tyrosine; O-Phospho-L-serine; L-Threonine; L-Phenylalanine; L-Glutamic acid; Methionine |
| map04978 | Mineral absorption | 0.023 | L-Asparagine; L-Threonine; L-Phenylalanine; Methionine |
| map04726 | Serotonergic synapse | 0.028 | Prostaglandin E2; Thromboxane B2; Prostaglandin D2; Prostaglandin J2; Prostaglandin A2 |
| map00290 | Valine, leucine and isoleucine biosynthesis | 0.035 | 3-Methyl-2-oxobutanoic acid; L-Threonine; 2-Isopropylmalic acid |
| map00380 | Tryptophan metabolism | 0.044 | Tryptamine; Kynurenic acid; Xanthurenic acid; L-Kynurenine; Quinolinic acid; Indole-3-acetic acid |
| map00750 | Vitamin B6 metabolism | 0.045 | Pyridoxamine; Pyridoxine; D-Erythrose 4-phosphate; 4-Pyridoxic acid |
| Colon | map04971 | Gastric acid secretion | 0.007 | Histamine |
Table 2
KEGG Enrichment of characteristic metabolites derived from liver and colon between compared treatments TSS vs Con
| Tissue | MapID | MapTitle | Pvalue | MetaIDs |
| Liver | map00970 | Aminoacyl-tRNA biosynthesis | 0.006 | L-Asparagine; L-Tyrosine; O-Phospho-L-serine; L-Threonine; L-Phenylalanine; L-Glutamic acid; Methionine |
| map04978 | Mineral absorption | 0.023 | L-Asparagine; L-Threonine; L-Phenylalanine; Methionine |
| map04726 | Serotonergic synapse | 0.028 | Prostaglandin E2; Thromboxane B2; Prostaglandin D2; Prostaglandin J2; Prostaglandin A2 |
| map00290 | Valine, leucine and isoleucine biosynthesis | 0.035 | 3-Methyl-2-oxobutanoic acid; L-Threonine; 2-Isopropylmalic acid |
| map00380 | Tryptophan metabolism | 0.044 | Tryptamine; Kynurenic acid; Xanthurenic acid; L-Kynurenine; Quinolinic acid; Indole-3-acetic acid |
| map00750 | Vitamin B6 metabolism | 0.045 | Pyridoxamine; Pyridoxine; D-Erythrose 4-phosphate; 4-Pyridoxic acid |
| Colon | map04971 | Gastric acid secretion | 0.007 | Histamine |
| (1) MapID: map ID of enriched KEGG Pathway. (2) MapTitle༚title of enriched KEGG Pathway. (3) Over Representation Analysis was implemented using the hypergeometric test to evaluate whether a particular metabolite set is represented more than expected by chance within the given compound list. One-tailed p values are provided after adjusting for multiple testing. (4) MetaIDs༚list of those input metabolites that participated in this KEGG Pathway. |
(1) MapID: Map ID of the enriched KEGG pathways. (2) MapTitle: title of enriched KEGG pathways. (3) Overrepresentation analysis was implemented using a hypergeometric test to evaluate whether a particular metabolite set was represented more than expected by chance within the given compound list. One-tailed p values are provided after adjusting for multiple testing. (4) MetaIDs: List of those input metabolites that participated in this KEGG pathway.
Moreover, microbial products that passed into the liver via the portal vein were significantly up- or downregulated associating with TSS treatment. Specifically, the relative abundances of salicylic acid, N-phenylacetylglutamine, N-acetyl-D-galactosamine, 2-isopropylmalic acid, pimelic acid and L-ergothioneine were significantly increased, while the relative abundances of Glu-Glu, glycoursodeoxycholic acid, glycolithocholic acid, theophylline, o-toluic acid, 5-aminopentanoate, pseudouridine and D-cysteine were significantly decreased (Table S2). Apart from microbial products, 38 metabolites changed with TSS treatment and were cometabolized by the microbiota and its host.
Metabolites derived from Liver were significantly correlated with those from colon
The effects of TSS on substance metabolism among liver and colon were characterized by a number of metabolites whom significantly changed in response to the TSS or antibiotics treatments (Fig. 6a), especially for the comparable groups TSS vs Con, Van + TSS vs Van, and Van vs GSW treatments. Microbial fermentation of dietary items could produce a myriad of metabolites which might later be absorbed by enterocytes and reach the liver via the portal vein (Fig. 6d). As a result, liver-derived metabolites are strongly correlated with colon-derived metabolites. Consistent with this view, we observed that colon-derived metabolites APPC, PMP, 4-MCD, TTPD, and 10-hydroxydecanoic acid were strongly associated with liver-derived metabolites D-proline, SDMA, spermidine, hexanoylcarnitine, proline, Ala-Leu, ADTF, OIELTD, Pro-Leu, and 1-methylxanthine (Fig. 6b). Moreover, the correlation network was dramatically altered by TSS when combined with vancomycin treatment (Fig. 6e). In particular, the liver-derived metabolite APPC, which was originally present in the colon metabolic fluxes, was positively correlated with creatine, β-nicotinamide mononucleotide, and phenylacetylglycine whereas was negatively correlated with N-oleoyl dopamine and sakuranetin. Additionally, the signaling pathways enriched by liver- and colon-derived biomarkers partially reflect the important impactions of TSS treatment on the microbiota and its host. TSS treatment had the most differential effects on metabolic pathways, paralleling with some effects on neuroactive ligand–receptor interactions and protein digestion and absorption (Fig. 6c). Expectedly, these TSS-related pathways were further extended by group Van + TSS which combined TSS with vancomycin treatment, and these impactions were defined by phenylalanine metabolism, purine metabolism, pyrimidine metabolism, taurine and hypotaurine metabolism, vitamin B6 metabolism, etc. (Fig. 6f).
The impact of TSS treatment on microbial cross-talking with their host
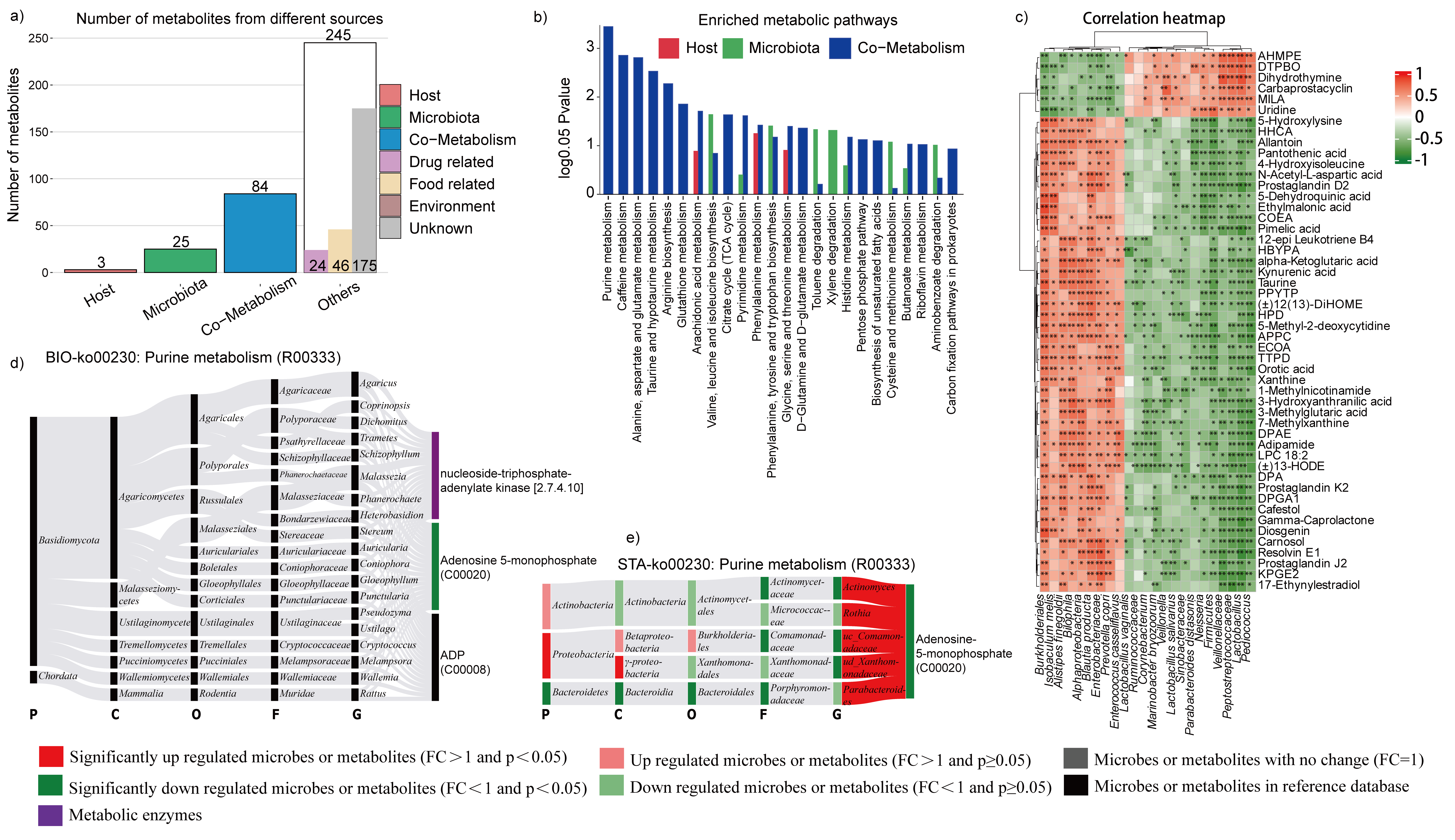
Among liver biomarkers that were obviously associated with TSS treatment, 5 metabolites were from the host, 15 were microbiota derived, 48 were cometabolized by both agents, and the others were specifically from the drug, food, environment or unknown sources (Fig. 7a). When we searched for the most enriched metabolic pathways according to metabolites derived from liver, bacteria, or both through metabolite pathway enrichment analysis (MPEA), we found that some cometabolism pathways shared by the gut microbiota and its host were characterized by aminoacyl-tRNA biosynthesis and vitamin B6 metabolism (Fig. 7b). Moreover, a cluster of bacteria, mainly including Coriobacteriaceae, Butyricicoccus pullicaecorum, Coprococcus, Mucispirillum schaedleri, and Anaerotruncus; unidentified F16; Lactobacillus vaginalis; Christensenella; Desulfovibrio; unidentified Flexispira; and Ruminococcus had positive correlation with hepatic metabolites. and another cluster had negative correlation with these hepatic metabolites, which included bacteria Corynebacterium stationis, Lactobacillales, Clostridium celatum, Veillonellaceae, unidentified rc4-4, Pasteurellaceae, Turicibacter, Pseudomonas, Peptostreptococcaceae, Enterobacteriaceae, Veillonella parvula, Barnesiella intestinihominis, Streptococcaceae, Prevotella, and Sutterella (Fig. 7c). The liver-derived metabolites D-proline, SDMA, spermidine, hexanoylcarnitine, proline, Ala-Leu, ADTF, OIELTD, prolylleucine, and 1-methylxanthine were the top 10 metabolites that were significantly correlated with the top 5 colon-derived metabolites (Fig. 6b). As mentioned above, the most enriched cometabolism pathway was aminoacyl-tRNA biosynthesis. Afterwards, all the microbiota that participated in liver metabolites production were drawn by the BIO-Sankey network. As shown, the phylum Euryarchaeota and Candidatus Bathyarchaeota were associated with the production of O-phospho-L-serine, demonstrating that they were significantly downregulated (Fig. 7d). Furthermore, an STA-Sankey network was drawn to reveal the microbial taxonomy accounting for metabolic changes. As shown, the enteric microorganism had a critical role in aminoacyl-tRNA biosynthesis, which included upregulated genus Coprococcus downregulated genus Veillonellaceae, unidentified Peptostreptococcaceae, unidentified Enterobacteriaceae, and Corynebacterium (Fig. 7e).
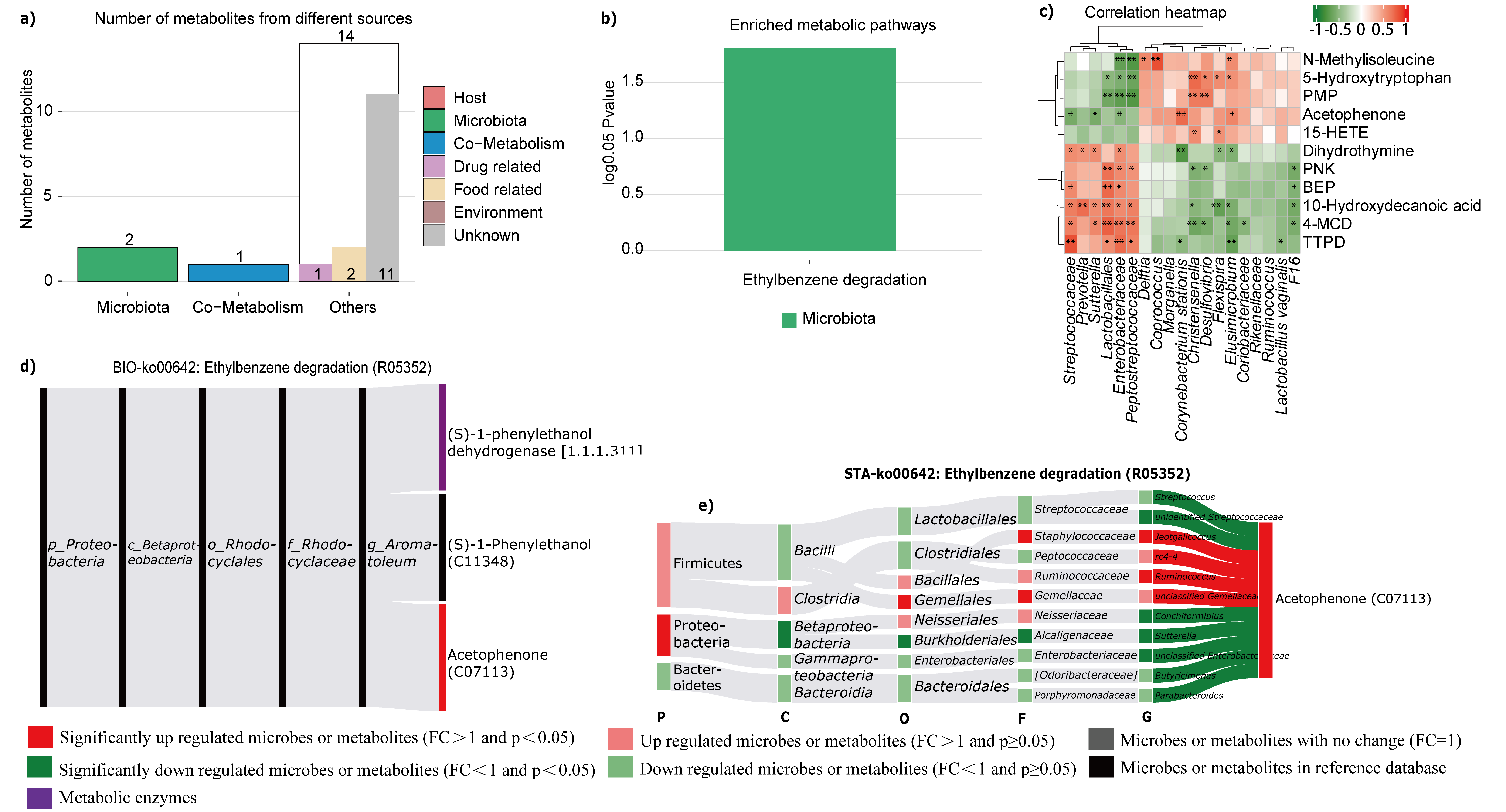
There were fewer metabolites derived from the colon had significantly altered with TSS than from the liver (Fig. S2a), and ethylbenzene degradation was the only metabolic pathway enriched in the microbiota (Fig. S2b). Moreover, the colon-derived metabolites N-methylisoleucine, 5-hydroxytryptophan, PMP, acetophenone and 15-HETE were positively correlated with microbe Delftia, coprococcus, Morganella, Corynebacterium stationis, Christensenella, Desulfovibrio, Flexispira, Elusimicrobium, Coriobacteriaceae, Rikenellaceae, Ruminococcus, Lactobacillus vaginalis, and unidentified F16, and were negatively correlated with Streptococcaceae, Prevotella, Sutterella, Lactobacillales, Enterobacteriaceae, and Peptostreptococcaceae. However, another group of metabolites, namely dihydrothymine, PNK, BEP, 10-hydroxydecanoic acid, 4-MCD and TTPD, had opposite correlations with the microbiota (Fig. S2c). Furthermore, according to the Sankey network (Fig. S2d, e), the abundance of significant downregulated microbes Streptococcaceae, Conchiformibius, Sutterella, and Enterobacteriaceae were negatively correlated with the abundance of acetophenone, which is involved in ethylbenzene degradation whereas the microbes Jeotgallcoccus and Ruminococcus, which were significantly upregulated, were positively correlated with acetophenone.
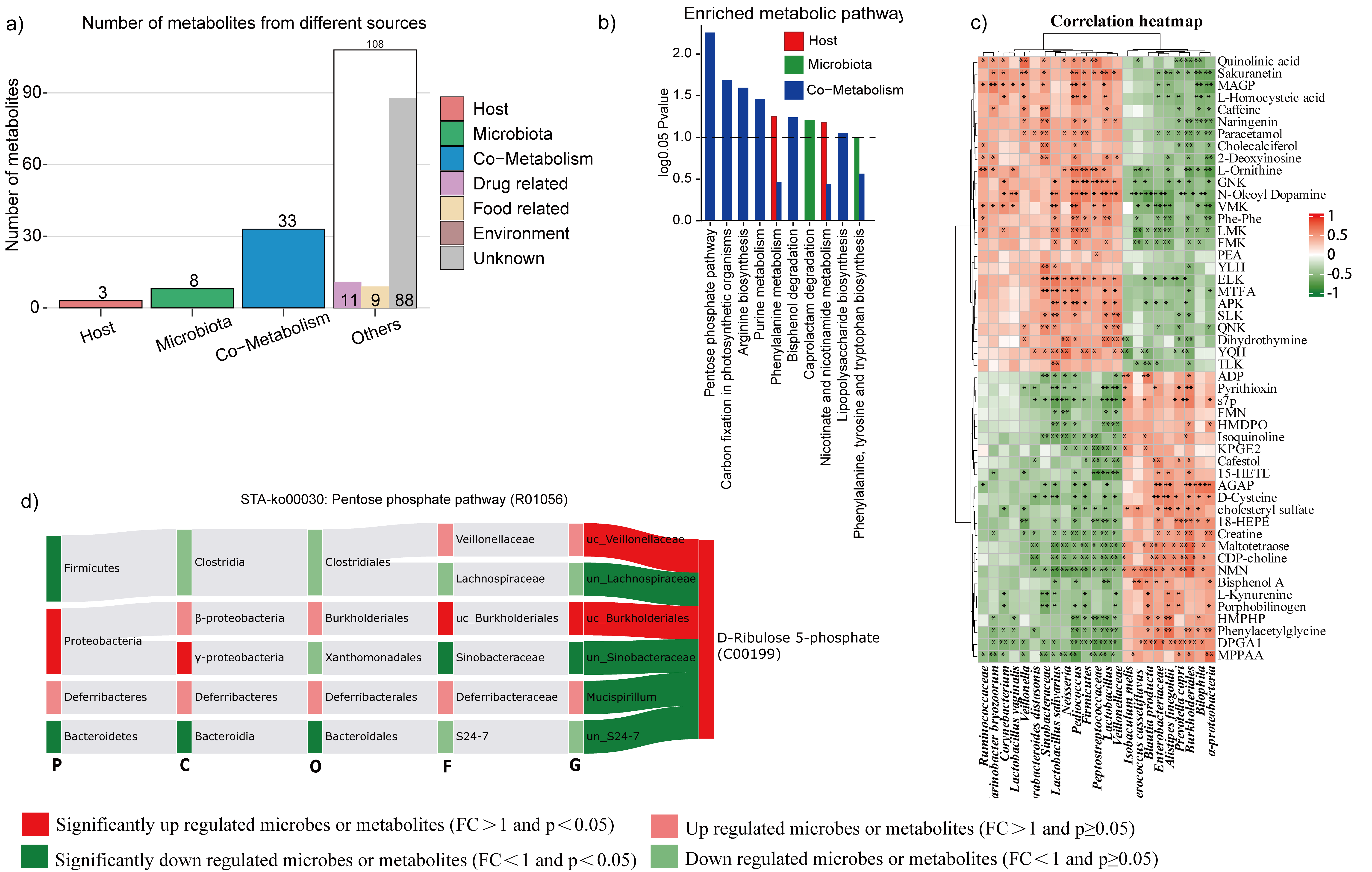
When intestinal microbes were disrupted by vancomycin, the cometabolic pathways and correlation maps were predominantly altered (Fig. S3a). Notably, the number of liver metabolites as well as enriched metabolic pathways (Fig. b) were obviously increased which were originally from both the microbiota and host-microbe cometabolism (Fig. S3a). And, the most effective pathway was the cometabolism of purines. Moreover, compared with TSS treatment, vancomycin administration changed greater numbers of liver metabolites that related to enteric microorganisms (Fig. S3c). Furthermore, we observed that the abundances of the genera Actinomyces, unidentified Comamonadaceae, and unidentified Xanthomonadaceae had significantly decreased and were positively correlated with the downregulated adenosine 5-monophosphate which was involved in purine cometabolism pathway (Fig. S3d, e). Moreover, when considering vancomycin treatment, the number of colon metabolites originating from the microbiota, host, and cometabolism was largely increased (Fig. S4a), as well as the number of significantly enriched metabolic pathways (Fig. S4b). Moreover, the total number of metabolites associated with microflora abatement increased markedly (Fig. S4c). Finally, analysis of the cometabolic pentose phosphate pathway demonstrated that the significantly increasing metabolite D-ribulose 5-phosphate was positively related to a significant increase in the abundance of the bacterial genus unidentified Burkholderiales and a decrease in the abundance of unidentified Sinobacteraceae (Fig. S4d).
{kind=link}
{kind=link}
{kind=link}
{kind=link}