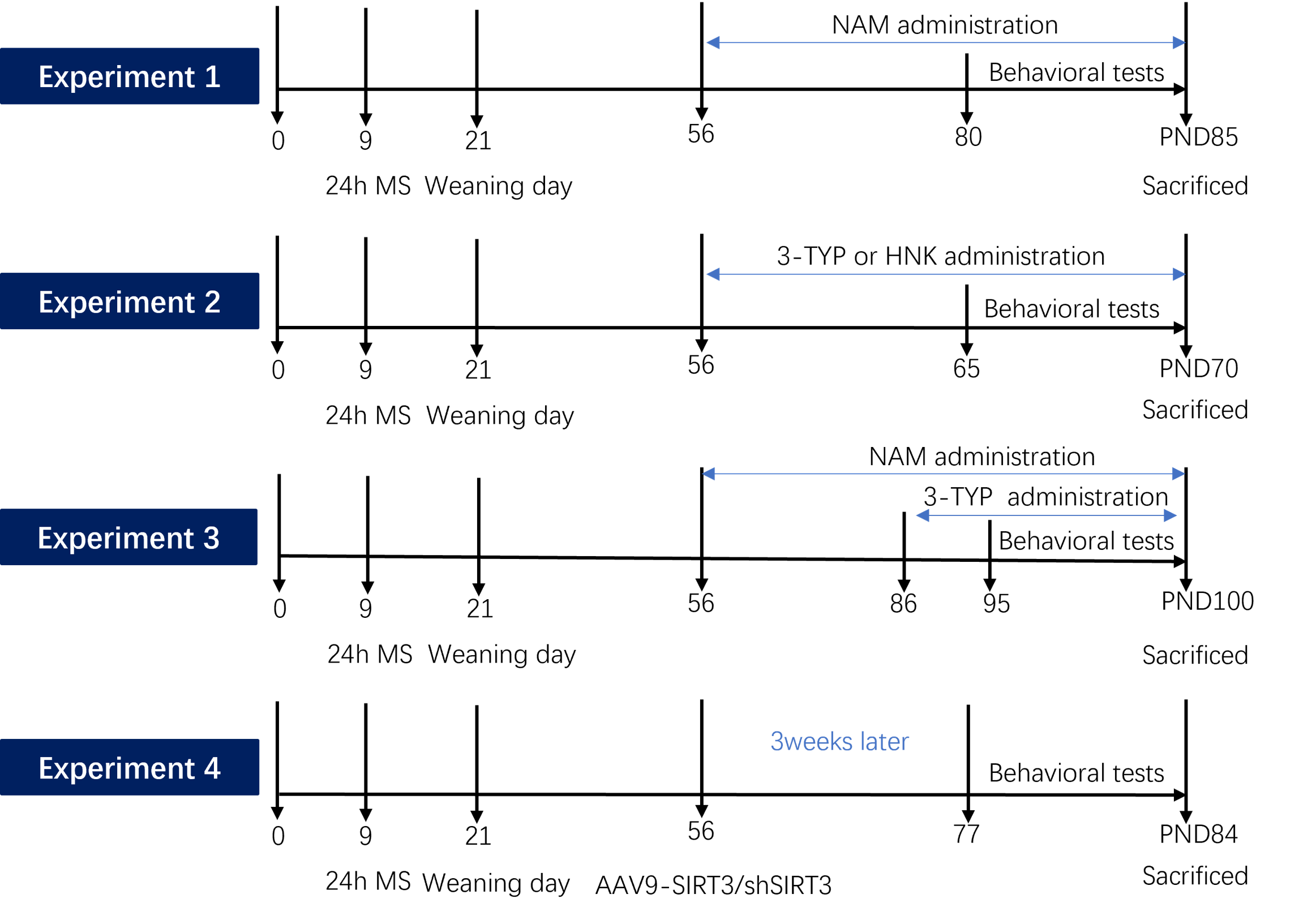
The main finding of this study was the identification of the involvement of the mitochondrial NAD+/SIRT3 axis in the pathophysiological mechanisms associated with schizophrenia. The study also provided a mechanistic account of the neurobiological intermediaries from the organelle to the cellular and synaptic levels. We observed alterations in the mitochondrial NAD+/SIRT3 axis in the hippocampus, reduced hippocampal neuronal spine density, impaired LTP in the CA1 region, and cognitive behavior deficits in the MS rat model. Furthermore, our results demonstrated that supplementing NAD + or activating/overexpressing of SIRT3 can restore hippocampal neuronal synaptic plasticity and mitigate cognitive impairment associated with schizophrenia in MS rats. Conversely, inhibiting or knocking down SIRT3 activity can lead to deficits in hippocampal neuronal and behavioral phenotypes. Our data strongly suggested that the NAD+/SIRT3 axis could be a crucial therapeutic target for schizophrenia and its associated cognitive deficits.
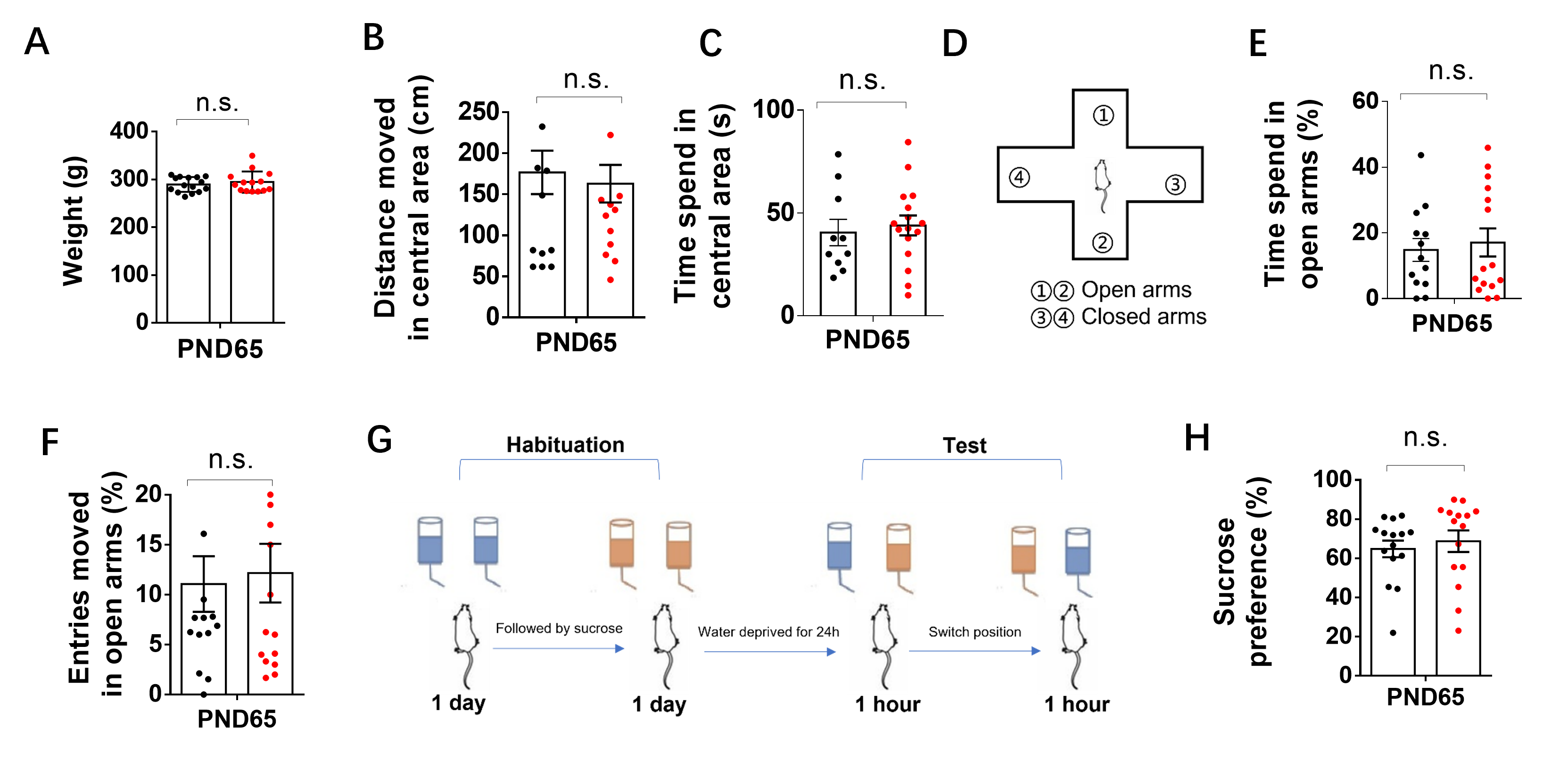
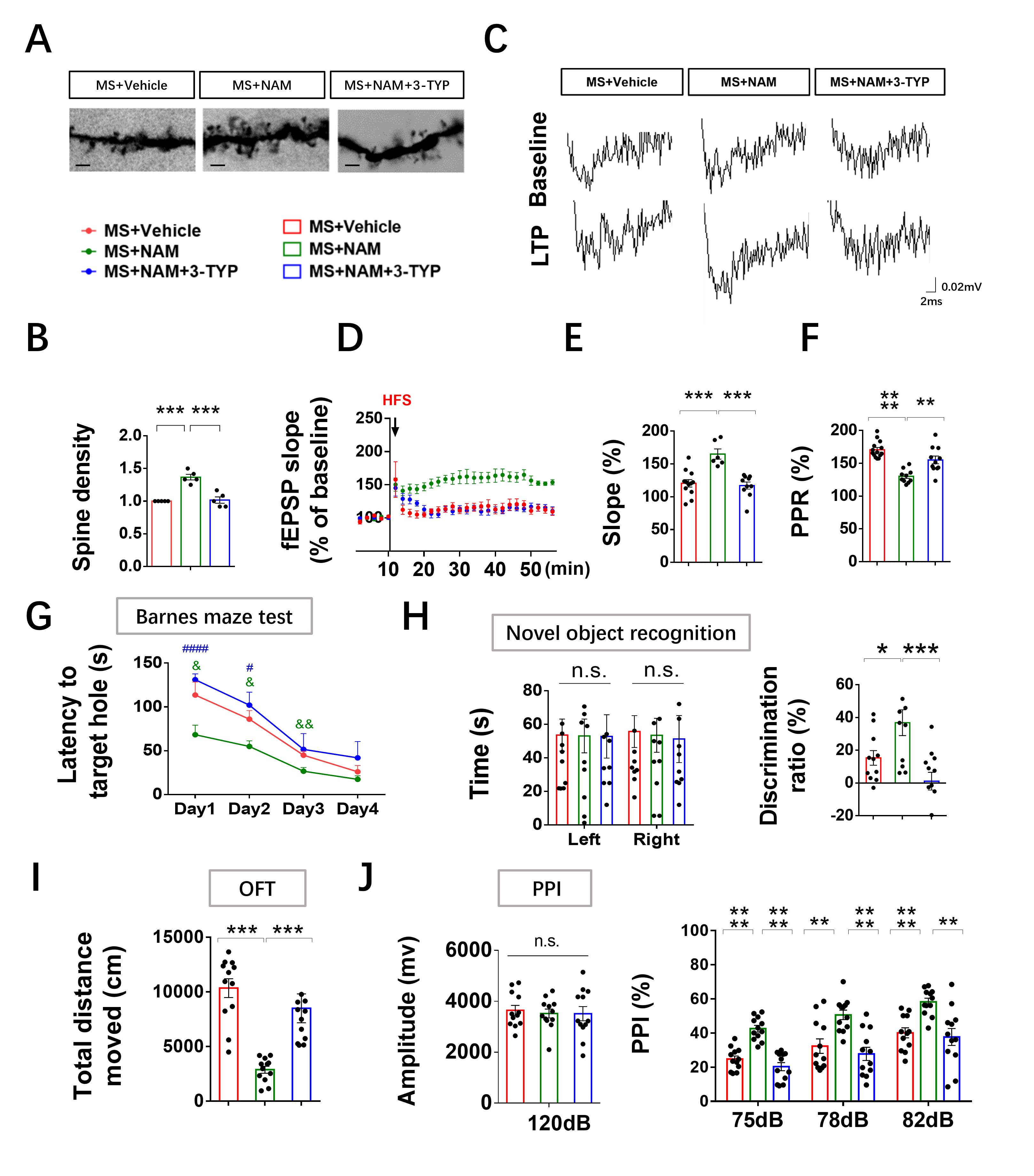
Mitochondrial dysfunction is a commonly reported phenomenon in schizophrenia [38, 39]. Early life stress can induce various behavioral changes [29–32, 40] and lead to alterations in gene expression and metabolism of mitochondria in different brain regions [41, 42]. However, the specific causal role of mitochondrial mechanisms in stress-induced behavioral changes remains unclear. MS, a widely-used and well-documented rodent model, has been extensively reported to induce schizophrenia-related behaviors and cognitive deficits in adulthood [29–32, 40]. Our findings supported these studies by demonstrating cognitive deficits in MS animals using the novel object recognition test and Barnes maze tests. The reductions in PPI further confirmed the validity of the schizophrenia model, as PPI impairments are observed in various neuropsychiatric disorders, including schizophrenia [43, 44], and are commonly used as behavioral assays in animal models of schizophrenia [45, 46]. Administration of NAM (a precursor of NAD+) normalized cognitive deficits, spontaneous activity, and impaired sensorimotor gating induced by MS. In addition, the SIRT3 activator or overexpression of SIRT3 attenuated most abnormal behaviors caused by MS. Meanwhile, the SIRT3 inhibitor or knocking down of SIRT3 led to spontaneous activity increase, abnormal spontaneous activity, PPI and impaired cognitive behaviors in adult control and NAM-treated MS rats. Therefore, a distinct contribution of our study was to have identified a novel role for the mitochondrial NAD+/SIRT3 axis in the regulation of early life stress-induced variation in behavior.
Spine remodeling, an important biological process shaping brain connectivity, has also been suggested to underlie complex behaviors and cognitive functions, such as learning and memory [47]. Reduced spine densities and elevated immature spines on the subiculum and CA1 regions have been previously found in patients with schizophrenia [48, 49]. The involvement of mitochondrial function in dendritic and spine complexity has been previously acknowledged for early development [13, 14, 17–19] and neurodegeneration [50, 51]. Our study showed that early life stress induced a reduced dendritic spine density in hippocampal neurons at the adult stage. Reduced dendritic spine numbers may induce reduced postsynaptic transmission, which may cause a significant reduction in LTP induction and LTP maintenance [52]. Consistent with previous study, our results showed that early life stress induced impaired LTP in these neurons in the hippocampal CA1 region. However, the mitochondrial contribution to molecular mechanisms regulating spine stabilization and maturation have not been fully understood.
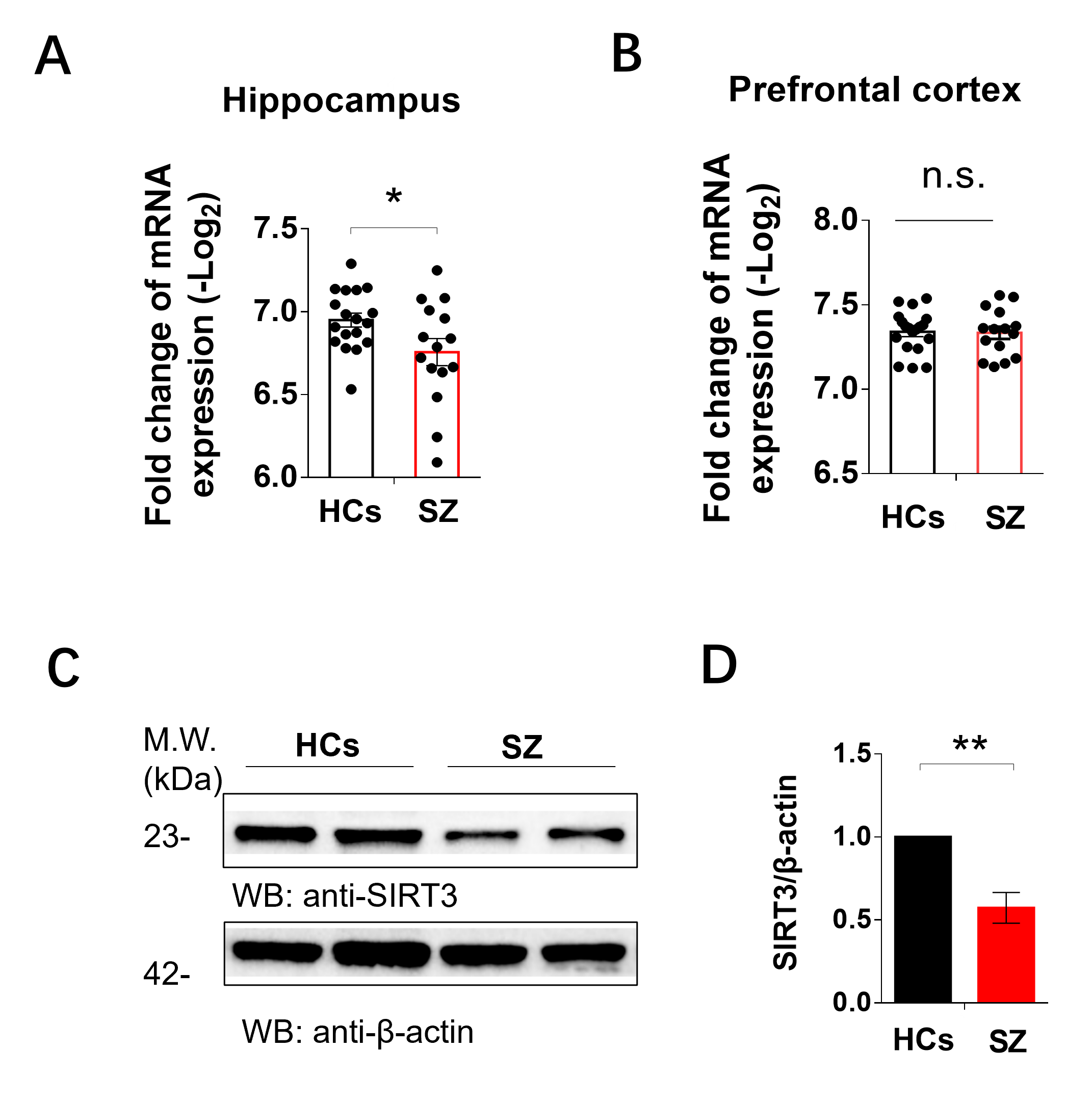
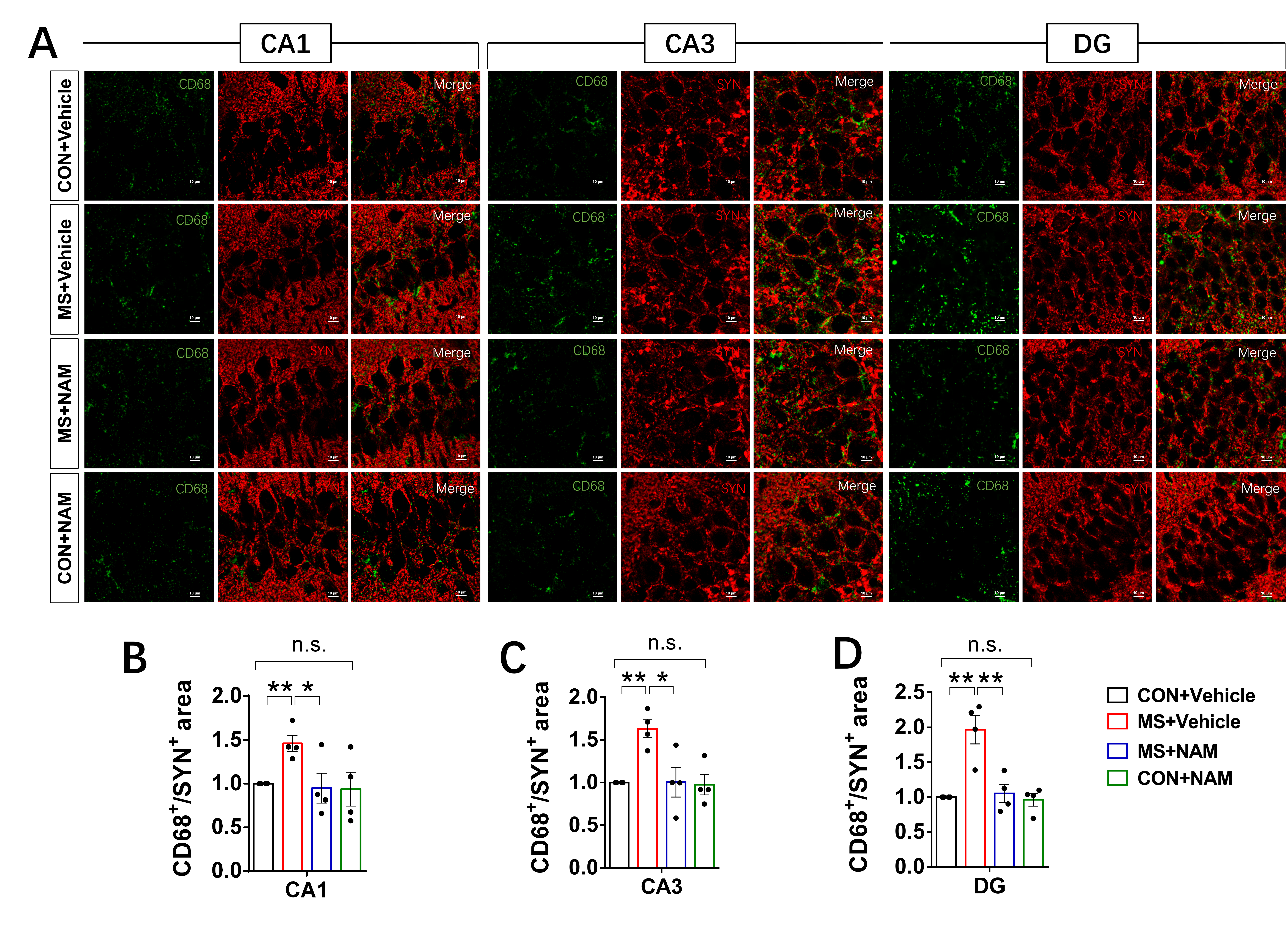
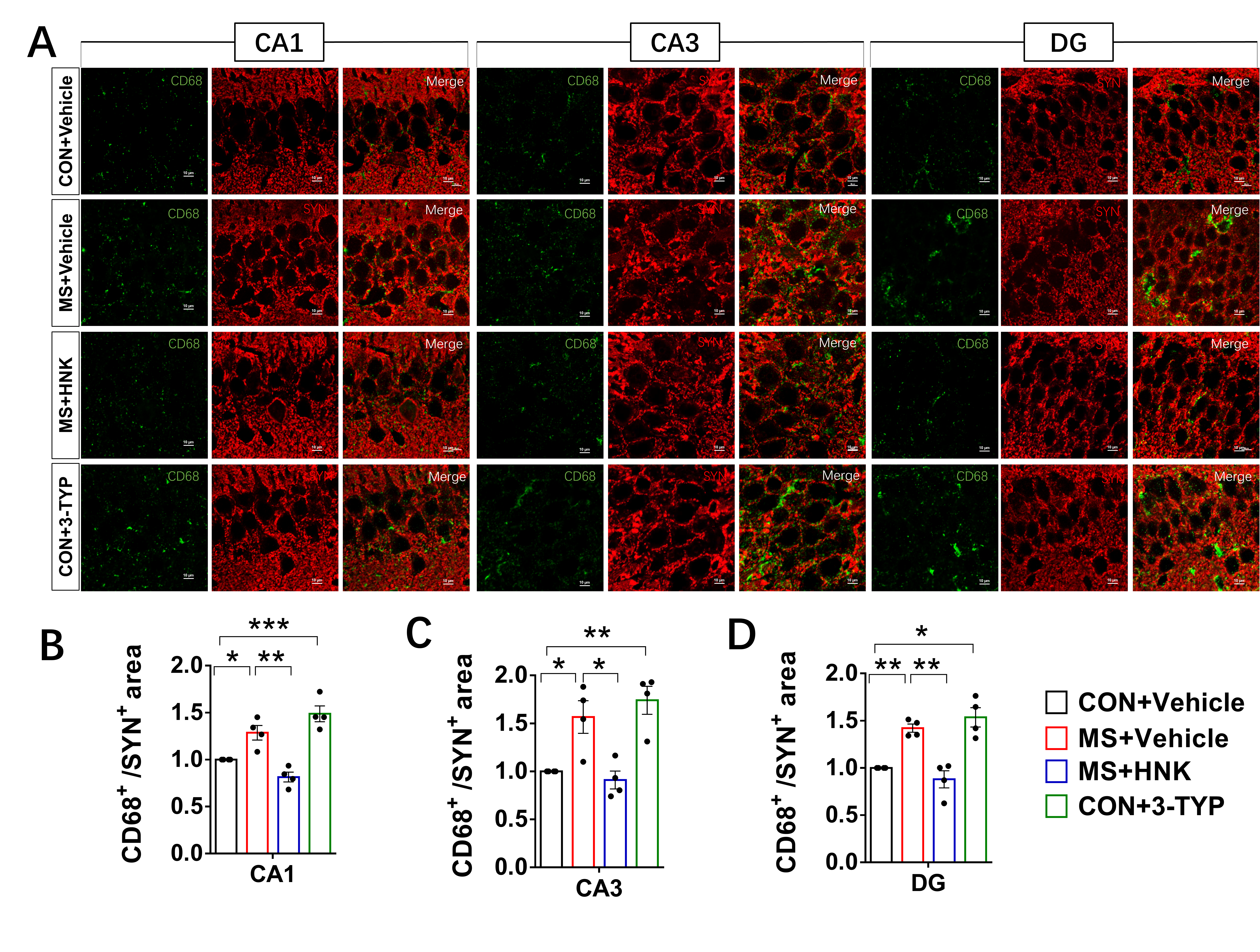
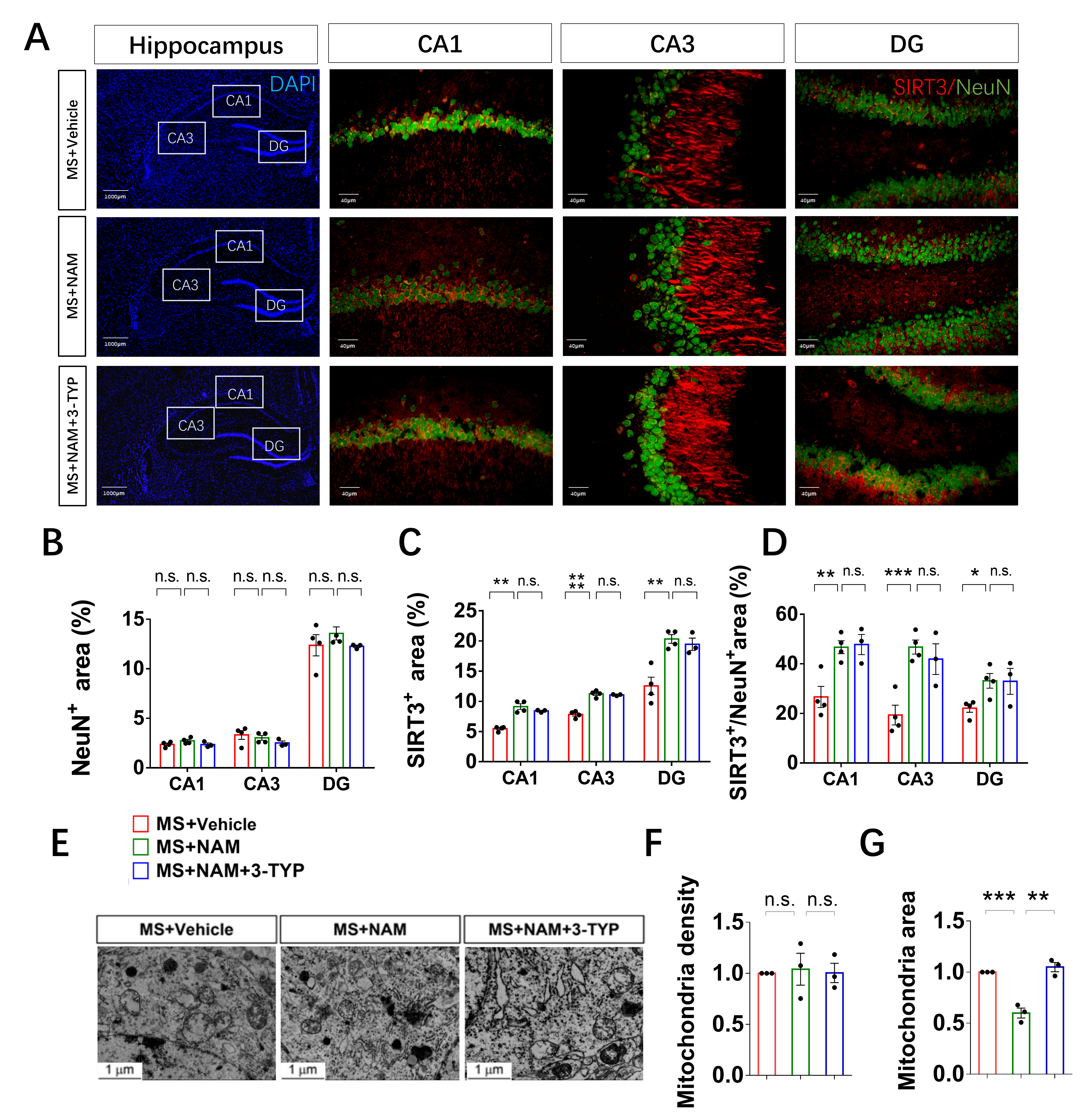
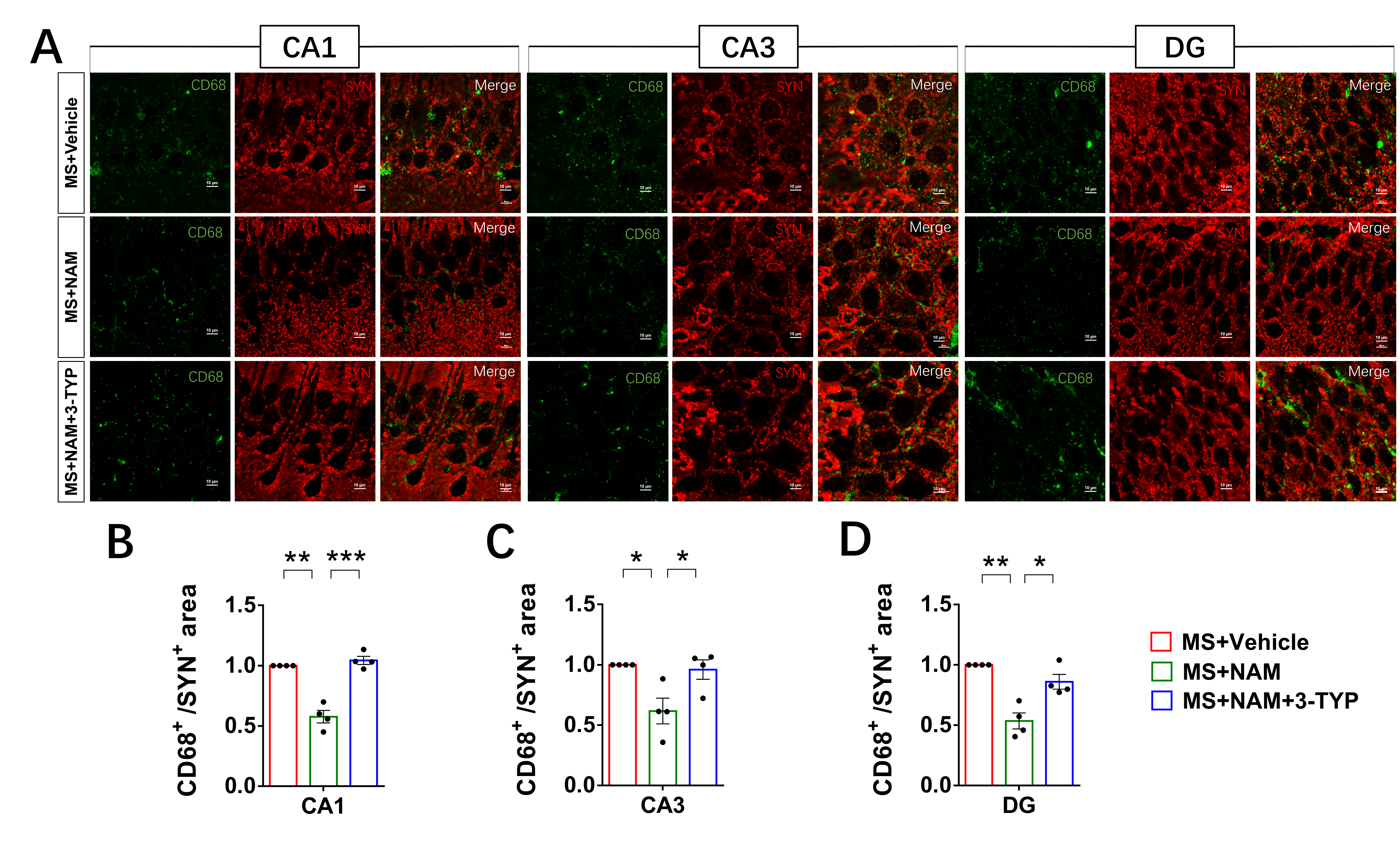
SIRT3, the primary mitochondrial NAD+-dependent protein deacetylase, plays a crucial role in maintaining mitochondrial redox homeostasis by regulating the function of electron transport chain complexes I and III, thus preventing ROS generation within the mitochondria [53, 54]. Our previous study has showed that SIRT3 inhibitor could induce ace-SOD2, ROS increase and mitochondria damage in the HT22 cells [21]. In this animal study, our results demonstrated that MS led to a decrease in SIRT3 levels in hippocampal neurons compared to control animals. Additionally, we observed mitochondrial swelling in the hippocampal neurons of MS rats. Studies suggest that mitochondria with a more rounded shape are often associated with less efficient bioenergetics [55, 56] and ATP levels [57]. Additionally, we observed an increase in microglial contacts with dendrites and microglial phagocytosis of spines under early life stress.
Microglia in the developing brain are highly mobile phagocytic cells with remarkably adaptable and versatile characteristics [58]. Apart from their notable functional adaptability, these cells are distinguished by a very low activation threshold, enabling them to carry out surveillance and scavenging functions effectively [59]. Studies suggest that the functional behavior of microglial cells may be influenced by neuronal activity [60]. Therefore, we hypothesized that alterations in the NAD+/SIRT3 axis within hippocampal neurons may contribute to the excessive engulfment of spines by microglia, leading to impaired LTP in the CA1 region and cognitive deficits in MS rats. To validate this hypothesis, we administered NAD + to adult MS rats. Our findings demonstrated the restoration of cognitive deficits, hippocampal neuronal synaptic plasticity, mitochondrial morphology, and NAD+/SIRT3 axis function in MS rats. Additionally, in order to further confirm the pivotal role of the NAD+/SIRT3 axis in regulating synaptic plasticity, we modulated the activity of SIRT3 in the animal models. Remarkably, in line with our hypothesis, the administration of a SIRT3 inhibitor to control and NAM-treated MS rats resulted in deficits in mitochondrial morphology, as well as in hippocampal neuronal and behavioral phenotypes. Activation of SIRT3 was able to reverse the mitochondrial, hippocampal neuronal, and behavioral phenotypes in MS rats. Furthermore, we conducted loss-of-function experiments through virus injection to investigate whether specific reduction of SIRT3 expression in hippocampal neurons replicated the behavioral and neuronal phenotypes observed in MS rats, which aligned with our initial predictions.
Our findings indicated that dysfunction of the NAD+/SIRT3 axis in hippocampal neurons leads to excessive microglial engulfment of neuronal spines, ultimately resulting in impaired synaptic plasticity. However, this intricate process involves various molecules, such as the classic complement cascade-dependent phagocytic signaling, transforming growth factor β, chemokine signaling, and brain-derived neurotrophic factor, which either promote or inhibit the elimination of specific synaptic connections [37, 61–65]. Further research is warranted to elucidate the precise mechanisms underlying the interactions between microglial cells and hippocampal neurons.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}