Widespread parasite resistance to artemisinin therapy is hampering malaria eradication programs, and more research into the host response to infection is needed to develop adjuvant therapies that minimize complications. [1, 2]. Malaria deaths reached 608,000 cases in 2022 with 249 million clinical malaria infections. Pregnant women and children under 5 years of age are most likely to develop severe malaria, with children < 5 years of age accounting for 67% of all malaria deaths [3]. Five parasite species cause malaria, and Plasmodium falciparum (P. falciparum) is the most virulent, responsible for 95% of malaria deaths and 200 million annual clinical infections. The life cycle of P. falciparum is complex, including sexual and asexual stages in female Anopheles mosquitoes and human hosts. When a female Anopheles mosquito infected with P. falciparum bites a person, it injects falciparum sporozoites into the skin. These sporozoites then travel to the liver and invade a hepatocyte, where they undergo a cycle of asexual replication. This produces a hepatic schizont containing numerous merozoites. The merozoites then enter the bloodstream and infect red blood cells (RBCs), developing from ring stages to trophozoites and then to schizonts. Once mature, the schizonts rupture, releasing more merozoites that continue the replication cycle. Finally, some of the parasites produce male and female gametocytes. The mosquito takes these up to continue the cycle [4].
Fatalities occur due to serious complications of malaria. These complications include cerebral malaria (CM), lung injury, renal failure, acidosis, and severe anemia [5–7]. The virulence of P. falciparum is attributed to the parasites’ ability to modify the erythrocyte surface to adhere and to evade the host immune attack. The major antigenic ligand found to be responsible for the cytoadhesive properties of the infected red blood cells (iRBC) are members of the P. falciparum erythrocyte membrane protein-1 (PfEMP1) family [8–9].
The pathogenesis of severe malaria is hotly debated; some researchers suggest that cytoadhesion is the overriding pathogenic mechanism, whilst others believe that inflammatory processes are more important. An accumulating body of evidence indicates that vascular endothelial dysfunction is also important and could be the interface between cytoadhesion and inflammation [9–12]. We concluded that establishing the causal role of any single mechanism in severe malaria in human is difficult. That’s why we planned to investigate more deeply into the host-parasite interaction mechanisms.
In the human host, RBCs and endothelial cells (ECs) are the primary cells that interact with parasites during P. falciparum infection. During this host-parasite interaction, biological material is exchanged. This includes the transfer of human microRNA (miRNA) between iRBCs and other non-infected cells within the human host, including non-infected RBCs, ECs, and immune cells [13]. Although miRNAs are known to control 60% of the human genes, but their role in malaria complications remains incompletely understood.
miRNAs
miRNAs are non-coding RNAs approximately 20 nucleotides in length, and are found in mammals, plants, and viruses. According to miRBase version 22, the human genome encodes ~ 300 true mature miRNAs, of which 1,115 are currently annotated [14]. An individual miRNA can repress tens to hundreds of genes, and miRNAs regulate nearly every cellular and metabolic process [15–17]. Since the discovery of miRNA in 1993, miRNA has been increasingly recognized as an essential post-transcriptional gene regulator [17]. Different steps of miRNA biogenesis occur in the nucleus and cytoplasm. In the nucleus, the corresponding region of the genome is transcribed by RNA polymerase II to produce primary miRNA (pri-miRNA) > 200 nucleotides in length. The DiGeorge Syndrome Critical Region 8 (Drosha/DGCR8) complex, which consists of RNase III family enzymes, crops the pri-miRNA into precursor miRNA (pre-miRNA) 70 nucleotides in length [18]. The pre-miRNA is then transported into the cytoplasm. The trans-activation responsive RNA-binding protein (TRBP)/dicer complex cuts the hairpin-like structure in pre-miRNA, processing pre-miRNA into a 14-21-nucleotide duplex mature miRNA structure comprised of two strands, the mature miRNA targeting strand and the miRNA passenger strand. mRNA target recognition occurs via binding of the short seed region (2–8 nucleotides) at the 5’ miRNA end (guide strand) and a partially or perfectly complementary region on the target mRNA 3’ untranslated region (UTR). This binding interaction represses translation of target mRNA and in some cases targets mRNA for degradation. The passenger strand was thought to be degraded, but recent research has shown that both miRNA strands have functional significance [19–20]. It was previously thought that miRNAs only negatively regulate target mRNA, but recent observations suggest that miRNAs can both repress and stimulate gene expression depending on various factors such as cellular conditions, sequences, and cofactors [21].
isomiRs
The discovery of miRNA isoforms, or isomiRs, was facilitated by high-throughput technologies such as deep RNA sequencing. IsomiRs differ from canonical miRNAs in length and/or sequence and are generated via RNA modifications catalyzed by enzymes such as deaminases and exonucleases. These molecules were initially hypothesized to be RNA-sequencing/mapping errors. However, subsequent studies demonstrated that small RNA-sequencing data contained a significantly higher percentage of untemplated nucleotide additions (%NTA) than the expected sequencing error rate calculated using small artificial RNAs. These studies confirm that canonical microRNA sequence modifications are physiological events that occur in vivo rather than experimental artifacts, which has been supported by the development of more advanced analysis algorithms. IsomiRs, like their canonical counterparts, have distinct functional roles. isomiRs bind Argonaute (Ago) proteins, as demonstrated by co-immunoprecipitation assays, and inhibit target gene expression, as demonstrated by in vitro luciferase assays [22–25].
Vascular endothelium
Approximately 96,000 km of blood vessels contained in the human body are maintained by ECs, a heterogeneous cell type that responds to signals in the microenvironment. ECs line the blood and lymph vessels except for the placenta, and form a physical barrier between the blood and tissue. ECs consist of a luminal membrane in direct contact with blood and circulating cells [26–27] and a basolateral surface. The basolateral surface is separated from the surrounding tissue by a basement membrane consisting of glycoproteins produced by ECs and anchored to their cell membrane. ECs perform numerous functions that depend on their location and activation status. These include maintaining vascular tone, controlling homeostasis, transporting hormones, and recruiting immune cells. ECs initiate and amplify the inflammatory response to vascular insults. ECs exhibit tissue type-specific heterogeneity. For example, tight junctions, absence of fenestrae, reduced pinocytic activity, and carrier-mediated transport systems are unique features of ECs that maintain the blood-brain barrier (BBB). The BBB protects the central nervous system from pathogens and toxic substances and restricts entry of antibodies and immune cells [28].
Endothelial shear stress
Vascular ECs sense hemodynamic changes and signals transmitted by the blood and respond by releasing vasoactive substances [29]. Shear stress affects EC morphology, electrochemical activities, and gene expression. Most importantly, NO release increases in response to shear stress, which is regulated by rapid activation of endothelial nitric oxide synthase 3 (eNOS) and upregulation of eNOS gene expression and transcriptional activation [30].
Under physiological conditions, endothelium-derived relaxation and contraction factors are in equilibrium. Vascular homeostasis is maintained with a slight bias in favor of vasodilation [29]. The endothelium is exposed to radial forces caused by intravascular pressure, tangential forces in the vessel wall caused by the balance between cell-cell contacts and vessel movement, and axial shear forces caused by the friction of blood flowing against the vessel wall. ECs are typically exposed to shear stress of 1–100 dyne/cm2 in the form of steady or pulsatile unidirectional shear. In culture, ECs are different from cells that grow in static conditions, forming a functionally confluent monolayer of cells. ECs can sense and respond to many stimuli, including the shear force of blood flowing through cell-cell junctions, heterotrimeric G proteins, primary cilia, caveolae, integrins, and the glycocalyx (GCX) [29–30].
Extracellular vesicles
Extracellular vesicles (EVs) are tiny lipid particles released by every living cell. They contain proteins, lipids, RNA, and DNA and can transport bioactive molecules. EVs also act as carriers that facilitate cell-to-cell communication, including antigen presentation and inflammatory activation [31, 32]. Recently, there has been a lot of interest in studying the biology and functions of EVs, particularly in immune regulation and host-pathogen interactions.
Vascular cell-pathogen interactions
Jambusaria et al. referred to the genetic signature of ECs as “postal codes” for organ-specific drugs. Identifying the differences in genetic signatures between ECs in different organs provides insight into the molecular underpinnings of EC heterogenicity [33]. Prior studies have explored the transcriptional landscape of brain ECs, and a smaller number of studies have evaluated the transcriptional profiles of lung ECs, but very few studies have explored the organ-specific heterogenicity of EC miRNA profiles. Lung ECs are in contact with the external environment due to their gas exchange function, and thus must initiate a rapid immune response in the event of infection. Lung ECs also facilitate the entrance of immune cells into the lungs to fight invading bacteria and viruses. Contrastingly, brain ECs prevent toxic molecules from entering the brain and maintain a much tighter barrier structure [27]. Understanding how ECs of different organs respond to stimuli contributes to drug design for infectious disease.
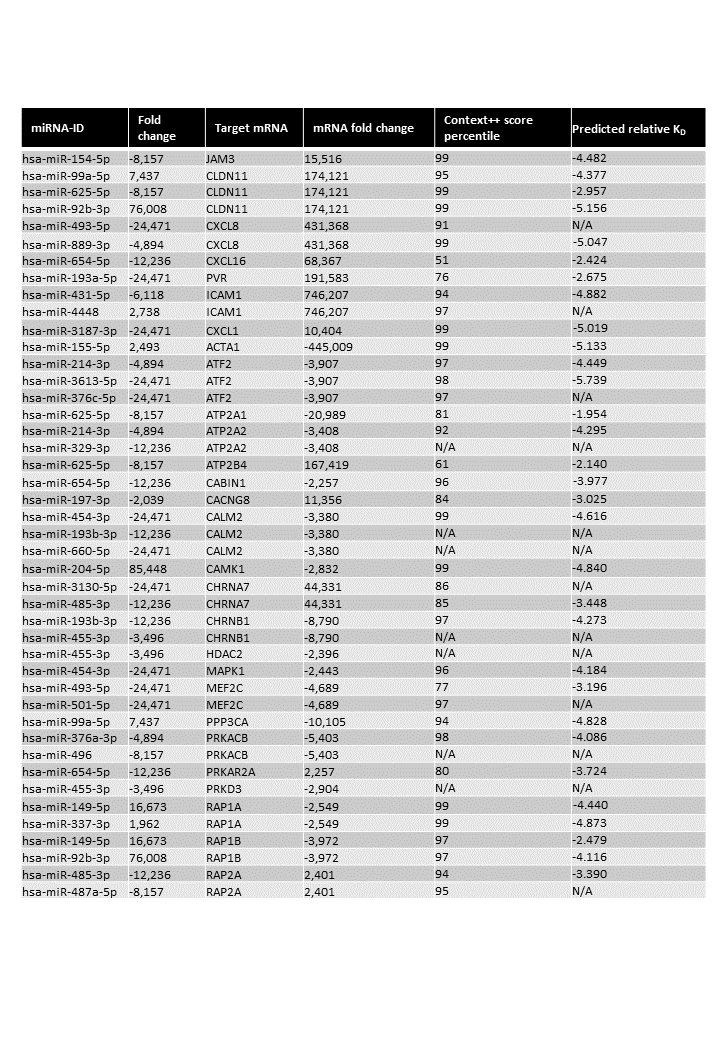
In the present study, we investigated intrinsic immune pathways in the brain and lungs ECs that might contribute to complications or protect the host. To pursue these scientific questions, we designed experiments to investigate the mRNA and miRNA profiles of primary human brain and lung ECs exposed to iRBCs. First, we compared the profiles of both ECs and EVs secreted by ECs to characterize the intrinsic heterogeneity between brain and lung ECs. Second, we exposed brain and lung ECs to authentic shear stress stimulation with similar stress levels to microvessels to create a physiologically relevant model. We subsequently evaluated the cellular response to shear stress and iRBC exposure to identify affected pathways and identify miRNAs that could potentially regulate these pathways. We demonstrated that shear stress activated multiple processes in brain ECs, including IL-8 signalling and strengthening of tight junctions. miRNAs differentially expressed in the ECs potentially controlled these pathways. Incubation with ring-stage iRBCs results in the activation of endocytic pathways in brain ECs. Contrastingly, in lung ECs, the most prominent activated pathway was the electron transport chain. In silico analyses identified that in the context of malaria exposure, endocytosis and electron transport pathways were potentially targeted by candidate miRNAs. The candidate miRNAs were significantly altered following 8 hours of co-incubation with ring-stage iRBCs at a shear stress of 1.5 dyne/cm2.
{kind=link}