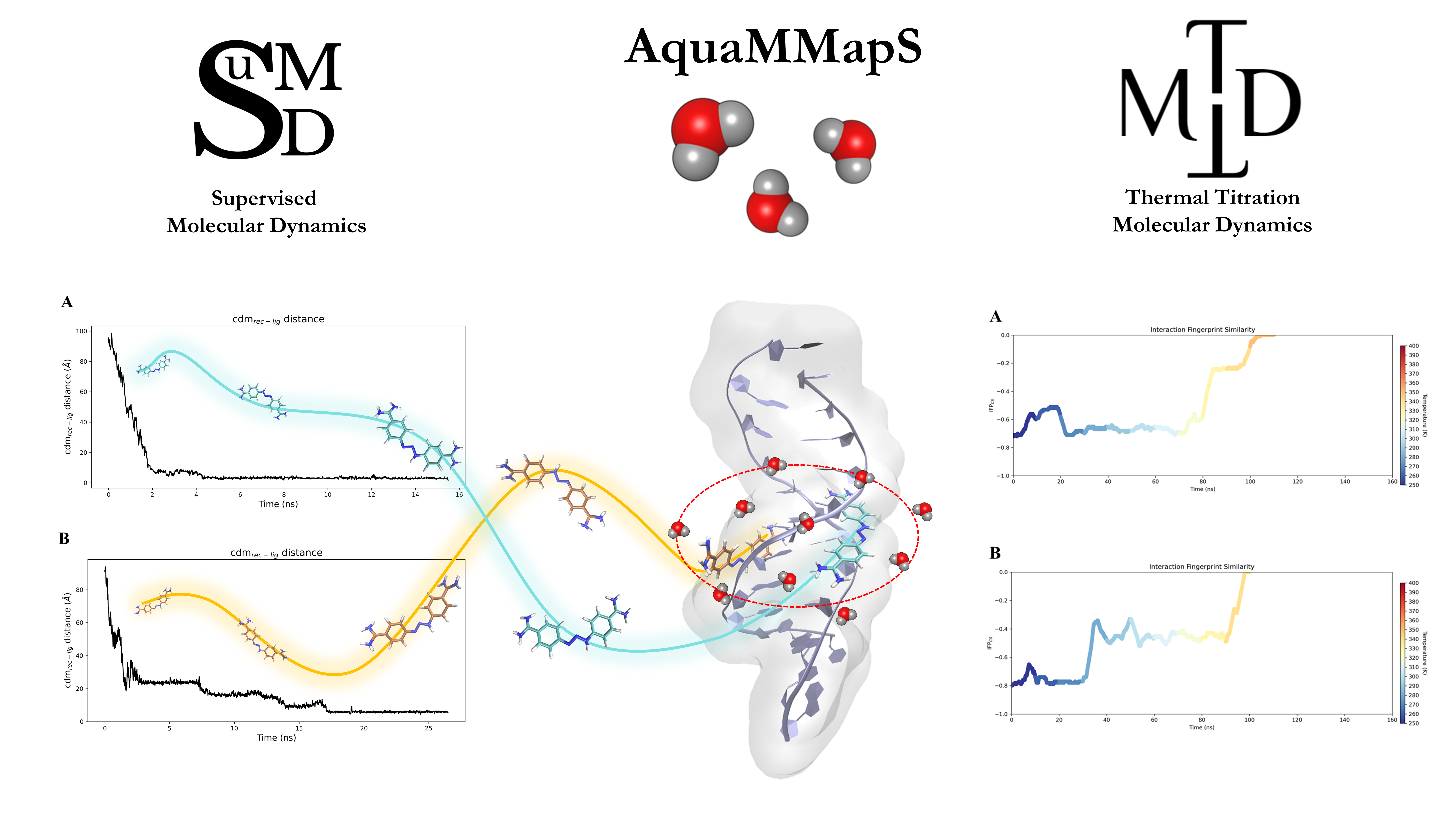
Starting with the structural information provided by the Berenil crystallography complex and the nucleotide sequences cataloged in the PDB under the 2DBE code, an attempt was made to generate a binding trajectory that could replicate the crystallographic binding mode, and its key interactions with the nucleic acid. To this end, Supervised Molecular Dynamics (SuMD) simulations have been conducted. SuMD is a well-established computational technique used for investigating unbiased receptor-ligand recognition pathways on the nanosecond timescale, by combining unbiased MD simulations and a tabu-like algorithm [10, 17]. This technique has been used in various studies to investigate the binding between several biomolecular receptors, including proteins, peptides, and RNA aptamers (available references). As depicted in Fig. 3 (Panel A), starting 95 Å away from the AATT nucleotide sequence, to avoid possible charge attraction effects, it was possible to explore the entire binding recognition in less than 16 nanoseconds (ns) of SuMD simulation.
The binding trajectory is rapid and linear. In fact, the ligand reaches the crystallographic binding site within the first 2 ns with favorable energy values. This is followed by a second phase of recognition and positioning extending up to 4 ns, where the ligand fully inserts into the minor groove. Despite the initial energetic disfavor observed during the first phase of recognition, the subsequent positioning within the minor groove is characterized by favorable and negative energetic interactions (Fig. 2 and Video S1).
By supervising the ligand proximity to the AT-rich binding site across the SuMD simulation, another stable binding mode at the level of AT-rich sequences was identified during the recognition phase. In this case, the ligand lies within the major groove displaying an alternative binding mode. The starting point of this simulation was identical to the one previously analyzed, but resulted in a different binding profile, observable in Fig. 3 (Panel B). In this case, the simulation runs for approximately 26 ns, and is characterized by a less direct approach path and several interactive steps along the trajectory.
The ligand undergoes a progressive reorientation during the binding trajectory towards the AT-rich binding site. Four predominant DNA-ligand populations are observable in the energetic profile reported in Fig. 4 and Video S1. The comparison of the of minor-groove and major-groove binding trajectories and their interactions analysis are available in Video S1 in the Supplementary information.
The final frame of the generated binding trajectories revealed favorable interactions with water molecules in both minor and major grooves. A comparison of these binding modes and their heat maps with the crystallographic ones is reported in Fig. 5. The water molecules highlighted in the reported binding modes of the last frame of the molecular dynamics (panels A and B) are also present at the crystallographic level. The heat maps illustrate the mediation of favorable electrostatic interactions with the ligand carried by the water molecule. This data further confirms the crucial arrangement of water during the dynamics, observable even in the deposited crystal, in which water bridges interactions between the ligand and the nucleic acid.
The interactive profile of the crystallographic binding mode (panel B) mirrors the one generated from the final frame of the dynamics in the minor groove (panel A), both in terms of electrostatic and hydrophobic interactions. For the major groove binding mode, the heatmap reveals reduced electrostatic interaction with the water molecule and, compared to the other two heatmaps (panel C), significant hydrophobic interaction differences, with several points of interaction missing.
In parallel with the exploration of the binding trajectories, we conducted a molecular dynamics study of the unbinding event using Thermal Titration Molecular Dynamics (TTMD) to evaluate the stability of a ligand-nucleic acid complex. This technique has been successfully applied to both protein and RNA targets [11, 12, 16]. For both compounds’ binding modes, five simulations were conducted, starting at 250 K, increasing the temperature until the binding mode was lost, resulting in the ligand exiting the binding pocket. The starting point for the molecular dynamic simulations was the last frame of the previously obtained SuMD run. The analysis of the trajectories is shown in Fig. 6. The comparison of unbinding trajectories and their interaction analysis are available in Video S2 in Supplementary information. For the minor groove-bound ligand (Fig. 6, panel A), the pose remains stable throughout the temperature gradient explored, for the large part of the simulation. The Interaction Fingerprint Similarity (IFPcs) values compared to the initial reference, starting at a value below − 0.7, remain constant throughout the temperature gradient explored until the ligand exits the binding site, at 320–330 K. There is only a first reassessment of the ligand in the first 20 ns of the simulation, with a slight loss of the IFPcs. This steady trend is also reflected in the ligand’s RMSD values, which remain stable during the simulation. On the contrary, the major groove-bound ligand (Fig. 6, panel B) shows a loss of interaction profile at lower temperatures. After 30 ns of simulation, the ligand loses its starting binding mode, indicating that it may have a shorter residence time, further confirming the preference for the minor groove. The reasons behind this propensity may be sought in a better shape complementarity as discussed in the original work of Professor Neidle, and in a steadier interaction between the nucleic structure and water molecules that can stabilize the binding mode. Considering that not only the nucleic-ligand interactions but also the solvent-mediated ones are crucial for a comprehensive understanding of both the binding and unbinding event, an insight into the stability of water molecules was carried out. For this purpose, in addition to the fingerprint similarity analysis, the AquaMMapS tool was used to map stable waters throughout the TTMD runs.
Figure 6 Interaction Fingerprint Similarity and RMSD values of minor-groove last frame of the SuMD simulation (panel A). Interaction Fingerprint Similarity and RMSD values of major-groove last frame SuMD simulation (panel B)
It is important to stress the fact that during the simulations and even in the equilibration phase, all the waters were not constrained. This approach facilitates the system’s proper solvation and, as a result, among the most stable waters, the ones occupying a “crystal-like” position, were obtained spontaneously.
Despite the rising temperatures and the exposure of the binding sites to the solvent, AquaMMapS revealed the persistence of the crystallographic water molecules identified along the unbinding trajectory during the Thermal Titration Molecular Dynamics (TTMD) simulation and also observable in the abovementioned last frame of SuMD simulation (Fig. 4 panel A and C). In the selected TTMD replica of the ligand in the minor groove, two stable water molecules were identified in the position occupied by the crystallographic one, exhibiting a B-factor value of approximately 21, as shown in Fig. 7 (panel A). Similarly, in the simulation of the ligand in the major groove, a stable water molecule was detected with a B-factor of approximately 25, as depicted in Fig. 7 (panel B).
The data highlights the crucial role of these water molecules as emphasized in Professor Neidle’s original study. To further characterize the role and location of these water molecules, an in-house Python 3.10 script was used to examine the dynamic profile of water molecules that were observed to interchange in the positions identified via AquaMMapS. This behavior can be examined in Fig. 7 (panels C and D). The analysis is specifically focused on counting distinct water molecules that interchange positions within the radius of a single water molecule at the chosen coordinates of the crystallographic waters. For each of these distinct water molecules, the number of simulation frames where they were detected was considered. The lower the number of distinct water molecules interchanging positions during the dynamics, and the more the number of frames where these water molecules are detected, the higher the hydration stability in that position. The results reveal that the count of distinct water molecules capable of interchanging positions in the minor groove is significantly lower compared to those in the major groove (~ 750 in the minor groove versus ~ 1200 in the major groove), with a notably increased frame count favoring the minor groove. This implies that these water molecules might be much more stable in the selected position in the minor groove than the major one.
{kind=link}