2.1 Materials
The solvents with HPLC grades were purchased from J.T. Baker (Philipsbur, USA). The LC-HRMS system consisted of an Ultra-High Performance Liquid Chromatography (UHPLC), model Infinity II 1290 (Agilent Technologies, Santa Clara, CA, USA), and a high-resolution mass spectrometer (HRMS) containing a quadrupole time-of-flight mass analyzer (QTOF, Impact HD) with an electrospray ionization (ESI) source (Bruker Daltonics, Bremen, Germany) (FAPESP 2014/50244-6).
2.2 Sponge material
The sample of sponges T. ignis (family Tedaniidae) was collected in high hydrodynamic coasts, in the intertidal zone, in São Sebastião, north coast of São Paulo state, Brazil, in the area of Praia Grande (23°49'23.76"S, 45°25'01.79" W) and Enseada do Araçá (23 No. 81'73.78 "S, 45 ° 40'66.39" W, São Sebastião, Brazil). Taxonomic identification was conducted using standard methods based on the morphological characteristics inherent to these sponges. Samples were immediately rinsed with saltwater, placed separately in containers with salt water, and transported to the laboratory in thermal boxes. In the laboratory, they were washed with Milli-Q water, weighed, stored at -20°C with proper identification, and then lyophilized.
It is important to notice that this study is registered in the National Management System of Genetic Patrimony (Sistema Nacional de Gestão do Patrimônio Genético, SisGen, registration number A4D38EE).
2.3 Extraction and sample preparation
The sample material of the sponge (3.96 g) was extracted with ethanol (EtOH) using Ika Ultra Turrax (T 25) for 5 min, 20.000 rpm at room temperature. The extraction process was performed in triplicate using 150 mL of EtOH as solvent. Thus, the extracted material was filtered and rotatory evaporation at 40 oC afforded crude extract dry materials (EETi, 56.7 mg).
The extract obtained was used to investigate antimalarial activity and for chemical fingerprinting.
2.4 LC-HRMS
The EETi sample (2.0 mg) was prepared using ethanol: methanol(20:80 v/v) in ultrasson for 10 min. Afterward, an aliquot of this extract mixture was diluted in ultrapure water at a final concentration of 500 µg/mL and resubmitted to ultrasson for 5 min. The mixture was centrifuged for 10 min at 12.000 rpm for analyses on HRMS (Gonçalves et al. 2020).
A XSelect® HSS T3 (2.5 µm particle size; 100 x 2.1 mm) (Waters, Milford, MA, USA) analytical column was used with a mobile phase composed of water (A) acetonitrile (B), with 0.1% v/v of formic acid which was added to both solvents. A linear gradient of 5 to 95% B in 20 min, keeping the 95% B composition for another 2 min, was used at a flow rate of 0.4 mL/min, 5 µL of injection volume, and a temperature of 40°C.
The ionization experiments were carried out in positive mode. The parameters used for mass spectrometry ionization source were as follows: nebulizer 4.0 bar, dry gas flow 9.0 L/min, dry heater temperature of 180 ºC, capillary voltage 4500 V, end plate offset 500 V, collision cell energy 10 eV, ion energy 5 eV, transfer time 50 and 90 µs, pre pulse time 6 µs and full-MS scan range 50-1300 m/z. The acquisition was obtained in auto MS/MS mode (cycle time 3 sec) in experiments with different collision energies of 20, 25, 30, 35, and 40 eV for all m/z analyzed (Gonçalves et al. 2020). Data acquisitions were carried out using the Data Analysis 4.0 software (Bruker Daltonics GmbH, Bremen, Germany), Compound Crawler Smartformula 3D, and GNPS. For compounds identification manual data curation was also necessary comparing results with literature.
2.5 Data Processing
The MS2 data acquired by LC-HRMS were converted into mzML (data files) using Data Analysis 4.0 (Bruker Daltonics, Bremen, Germany). Afterward, data was compressed using the program WinSCP client FTP and uploaded to the workflow into GNPS (http://gnps.ucsd.edu). The molecular networks were generated according to the standard protocol (https://ccms-ucsd.github.io/GNPSDocumentation/) available on GNPS platform (Wang et al. 2016).
After running a spectral clustering algorithm (MS-Cluster software), data sets were downloaded from GNPS pages into Cytoscape (version 3.8.2) together with MS2 features allowing for analysis of the network. In the molecular network, clusters comprised nodes joined by edges for compounds with similar product ions.
To generate molecular networking the mass tolerance was 0.02 Da for the precursor peaks, and MS2 fragment ion tolerance of 0.02 Da. A network was created where edges were filtered to have a cosine score above 0.65 and more than four matched peaks. Further, edges between two nodes were kept in the network if and only if each of the nodes appeared in each other's respective top 10 most similar nodes. The bordering of the molecular family was defined as 100. Finally, the spectra in the network were searched against GNPS' spectral libraries. All matches kept between network spectra and library spectra were required to have a score above 0.7 and at least six matched peaks.
The spectral libraries used for compound annotation were Mass Bank, NIST (National Institute of Standards and Technology), MoNa (MassBank of North America), and ReSpect. The data files deposited in GNPS can be accessed at https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=7398f47d17ed430d84b3e842a0c7d34d.
2.6 Maintenance of P. falciparum in vitro and SYBR Green assay
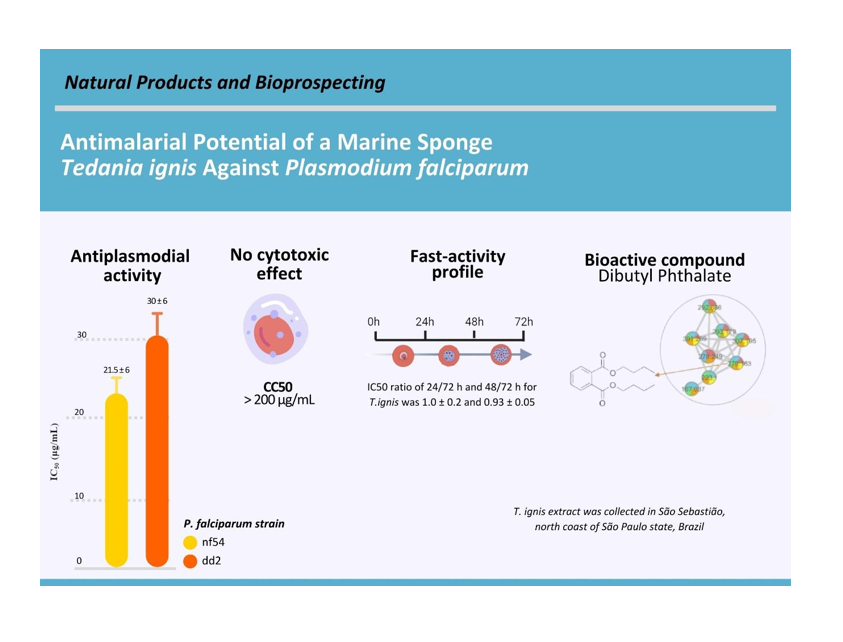
For the culture of the P. falciparum NF54 strain, the parasites were cultivated as described by Trager and Jensen (1976). They were maintained in RPMI culture medium supplemented with 0.5% albumax (GIBCO) at a hematocrit of 2% and parasitemia of 5%. For the experiments, the culture was synchronized with a 5% sorbitol solution at 37ºC for 10 minutes (Lambros et al. 1979). For the SYBR Green assay and evaluation of antiplasmodial activity, the hematocrit of the culture was adjusted to 2%, and parasitemia to 0.5% by adding O+ human erythrocytes. Twenty-microliter aliquots from serial dilutions, ranging between 100 µg/mL to 0.04 µg/mL, of the compounds under examination were prepared in a 96-well plate. Then, 180 µL of the culture with adjusted hematocrit and parasitemia were added per well. The plate was incubated at 37ºC in a humidified incubator with an atmosphere of 5% CO2 and 5% O2. In each plate, negative and positive inhibition control wells were added, containing parasitized RBCs without added compounds and non-parasitized RBCs, respectively. After incubation, the SYBR Green protocol [53] was applied to evaluate the inhibition of parasite growth. The intensity values obtained post-SYBR staining were normalized as percentage viability relative to both the positive and negative controls. The minimum inhibitory concentration of 50% (IC50) was obtained by analyzing dose-response curves plotted in GraphPad Prism 8.0 (Dery et al. 2015; Vossen et al. 2010).
2.7 Speed of action assay
To categorize the compounds based on their fast or slow-acting profiles, two protocols were simultaneously conducted, adapted from ( Le Manach et al. 2013).
The action duration of the compounds was assessed by preparing three identical plates (A, B, and C) with equivalent compound dilutions. Each plate was incubated with P. falciparum NF54 strain, wherein > 90% of the stages were in the ring form (synchronized), adjusted to 2% hematocrit and 0.5% parasitemia. The three plates were incubated with the inhibitor under growth conditions for durations of 24 hours (plate A), 48 hours (plate B), or 72 hours (plate C). Following the respective incubation periods, plates A and B underwent three washes with RPMI medium to eliminate the inhibitors and were subsequently incubated for an additional 48 and 24 hours, respectively. Plate C remained continuously incubated in the presence of the inhibitor throughout the entire period. Post-incubation, viabilities, and IC50 values for each plate were assessed using the SYBR Green I assay. The IC50 values from the three incubation times were compared to ascertain any significant differences in inhibitory potency (IC50) resulting from each duration (Fig. 1A).
2.8 Morphology assay
Associated with the speed of action assay, we conducted the morphology assay. In this analysis, parasites were incubated (2% hematocrit and 0.5% parasitemia, with > 90% in ring form) with 5 times the IC50 value of the inhibitor, under growth conditions for 24 hours. Afterward, the parasites were washed to eliminate the inhibitor and further incubated until reaching 72 hours. Blood smears were subsequently taken at 0, 24, 48, and 72 hours after incubation and stained with Giemsa to track parasite development. Parasitemia growth was quantified via optical microscopy at 0, 24, 48, and 72 hours after incubation and compared with the untreated control. This assay aimed to mitigate potential SYBr labeling errors that might occur in the speed-of-action assay model, as deceased parasites could be falsely identified by SYBr green at the 24-hour mark (Fig. 1).
2.9 Maintenance of cell culture
Two distinct cell lines were utilized: HepG2, derived from human hepatoma, and HEK 293, derived from human embryos. These cell lines were acquired from the Cell Bank of Rio de Janeiro. They were cultured in RPMI medium supplemented with 10% fetal bovine serum, sodium bicarbonate, gentamicin, 2g/L glucose, and HEPES at pH 7.4. Specifically, the HEK-293 cells were maintained in RPMI culture medium with similar supplementation except for 1g/L glucose. The culture medium was refreshed every 2 days, and their morphology was observed using an inverted microscope until reaching the desired confluence for conducting the experiments.
2.10 Cytotoxicity with resazurin assay
Cytotoxicity protocol, as detailed by Céu de Madureira (2002), was conducted in duplicate using cells maintained in confluent cultures and subsequently washed with serum-free medium. Following the addition of 5 mL of 1x trypsin and incubated for 5 minutes at 37°C. The harvested cells were then suspended in a complete medium, and centrifuged, resulting pellet that was resuspended in 10% SBF. After cell counting, the cell suspension were distributed in 96-well microplates at a concentration of 3x106 cells/100 µL/well and transferred in a CO2 incubator at 37°C overnight to promote adherence. Next, 100 µL of complete medium with different concentrations of the compound (T. ignis), ranging from 200 µg/mL to 0.195 µg/mL, was added to each well. After 72 hours, 40 µL of Resazurin solution was introduced into each well followed by 4-hour incubation, with the reduction of Resazurin resulted in a color change from dark blue to pink. The microplates were then analyzed using a spectrophotometer with 570 nm and 630 nm filters. The cytotoxic concentration at 50% (CC50), indicating the concentration where 50% of viable cells were observed in the presence of the test molecules and the control antimalarial, was determined by comparing it with cell cultures grown without the molecules, considered as exhibiting 100% growth.
2.11 Statistical analysis
The concentration and results of the IC50 and CC50 were evaluated based on the equation of the curve obtained by plotting the % of parasitemia regression vs the log of the concentration of extract. The average IC50 and CC50 were compared using the program GraphPadPrism 8.0.1.
{kind=link}