Study design
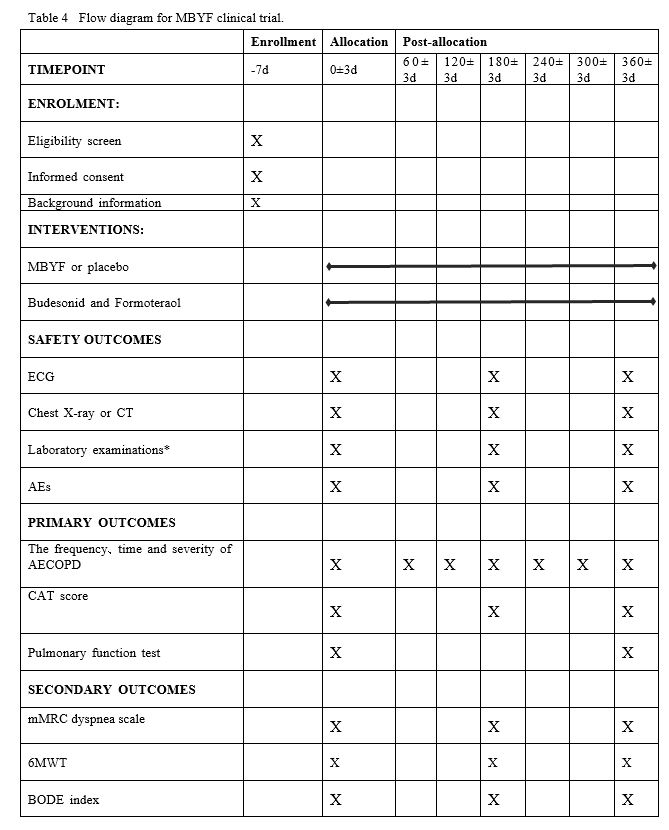
This is a multicentre, randomized, double-blinded, placebo-controlled, parallel-group clinical trial. We will rigorously follow the latest Consolidated Standards of Reporting Trials (CONSORT 2017) for Chinese herbal Medicines recommendations(39), and compile with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. Standard Protocol Items: Recommendations for Interventional Trials and 2013 statement for herbal interventions(40). Totally 300 participants diagnosed COPD will be enrolled in the trial, which consists of a 7-day screening period and a 360-day treatment period with every 60-day follow-up. In addition to routine medications, patients will be given 10.24g MBYF or placebo in granule form two times per day for 360 days. The participant’s flowchart is briefly illustrated in Figure 1.
Participant recruitment
Consecutive patients undergoing pulmonary function tests(PFTs) for a clinical indication will be evaluated for the entry into the study. Inpatients and outpatients at the ten participating hospitals will be screened according to the inclusion and exclusion criteria by two experienced respiratory physicians separately. Table 2 shows the cooperative hospital recruiting patients. The participants will be enrolled in the trial only when confirmed by two respiratory physicians at the same time. Also, recruitment advertisements of the study will be posted on webpages and notice boards in ten participating hospitals and resident communities. It includes a brief description of the subjects needed, the medicines, medical examinations and the ways to participate in this study. For those people who are ineligible or decline to participate, we will record the basic demographic information and reasons for non-participation. The first volunteer has been recruited on January 14, 2020 in Xinjiang. Due to COVID-19, we expect to complete the recruitment process around December 2020 and report the results as soon as possible.
Diagnostic criteria of COPD
The diagnosis of COPD will refer to the GOLD science committee report 2019. According to the comprehensive analysis of clinical manifestations, risk factor exposure history, signs and laboratory examinations and other data. Consider the main symptoms of COPD as chronic cough, sputum and/or dyspnea and risk factor exposure history. The incomplete reversible airflow limitation is a necessary condition for the diagnosis of COPD. The gold standard for COPD is FEV1/FVC <70% after using bronchodilators. Anyone with a history of smoking and/or exposure to environmental occupational pollution and/or cough, sputum or difficulty breathing, will be screened by PFTs. Chest x-ray examination helps determine the degree of the hyperinflation of the lungs. Exclude bronchial asthma, bronchiectasis, congestive heart and other lung diseases such as heart failure and tuberculosis.
Diagnostic criteria of TCM syndrome
TCM syndrome of ‘Fei_Shen_Qi_Xu’ will refer to ‘‘Clinic terminology of traditional Chinese medical diagnosis and treatment-Syndromes, GB/T 16751.2-1997’’. Table 3 shows the diagnostic criteria of ‘Fei_Shen_Qi_Xu’. If patients have two of the primary symptoms and two of the secondary symptoms, TCM syndrome of ‘Fei_Shen_Qi_Xu’ is determined.
Inclusion criteria
(1) Patients clinically diagnosed as COPD with airflow limitation: a post-bronchodilator fixed ratio FEV1/FVC < 0.7, 30% ≤FEV1 < 80% predicted.
(2) Patients without respiratory tract infection and acute exacerbation of COPD (AECOPD) in the past four weeks;
(3) Patients with TCM syndrome of ‘Fei_Shen_Qi_Xu’;
(4) Patients with severe exacerbation history: ≥2 or ≥1 leading to hospital admission in the past year;
(5) An age limit ranged from 40 to 85 years;
(6) Provision of written informed consent by participants or surrogates voluntarily.
Exclusion criteria
(1) Airflow obstruction caused by asthma and other non-COPD;
(2) Patients with lung transplantation or pneumonectomy;
(3) Patients needing long-term oxygen therapy (daily oxygen inhalation time ≥12 hours);
(4) Patients with a history of chronic alcoholism or drug abuse;
(5) Patients with the standard treatment of oral glucocorticoid at present;
(6) Patients with the treatment of pulmonary rehabilitation;
(7) Patients accompanied by serious diseases of heart, liver and kidney;
(8) Patients with a malignant tumour, leukaemia, aplastic anaemia, myelodysplastic syndrome, thrombocytopenia, multiple myeloma and other haematological diseases;
(9) Patients who are allergic or intolerant to the study of drugs;
(10) Participate in other clinical trials in the past four weeks;
(11) Pregnant women or breastfeeding women;
(12) Other.
Removal, dropout and termination criteria
Participants not taken medication during the treatment period will be removed. Participants can voluntarily drop out at any time during the trial. Eligible subjects failing to complete the observation period will be considered as dropout cases regardless of the time and reason. Reasons for dropout will be recorded in the case report forms(CRFs), and the last data recorded for these participants will be included in the data analysis. The trial will be suspended if serious adverse events (SAEs) relevant to the MBYF occur, or the participant decides to join in another clinical research project in terms of COPD, or demonstrates hypersensitivity towards MBYF, such as abnormal thirsty, stomach ache and diarrhoea, or suffers from the acute life-threatening disease, or with a combination of drugs, especially the drugs that affect the efficacy and safety of the MBYF.
The whole research will be terminated in the following circumstances: (1) masking of the randomization fails; (2) unblinding rate exceeds 20% of the sample size; (3) assessments of all follow-up are completed.
Randomization and blinding
According to the random number sequence generated by SAS, the patients will be randomly divided into either the MBYF group or the placebo group in a 1:1 ratio, using SAS to achieve computerised randomisation in blocks of 4, stratified by center. We set the blind code in the case for remaining blind when the patients have adverse effects. The random code and blind code will be conducted by a “third party” independent of the study using opaque envelopes. The envelopes will be sealed and shuffled and the assignment records will not be disclosed until the end of the study. To ensure the implementation of the blinding method, MBYF and MBYF placebo will be made in the same shapes, smells and tastes. Trial participants, care providers including attending physician and nurse, outcome assessors including PI and sub-PI, and data analysts will be blinded after the assignment of interventions.
Procedure for unblinding
If there is sevea re adverse event, which impedes the progress of the trial and the selection of the treatment measures, urgent unblinding can be carried out. During the process, all the researcher, sub-PI, and clinical supervisors should take part. The local administrative unit should be informed within 24h. The reason, time, and place of unblinding should be recorded in detail and all the records should be signed off.
Interventions
Budesonide formoterol powder inhaler (160 μ g / 4.5 μ g, once in the morning and evening, 2 inhalations each time) is provided as a conventional treatment to all participants according to the GOLD for COPD. MBYF or placebo( 10.24g/bag, one bag at a time, two times per day, 360 days) is provided as adjunct medication randomly using an envelope. MBYF and placebo(contain 10% MBYF) are identical in both taste、appearance and package and produced by Huarui Sanjiu pharmaceutical industry in Shenzhen, China. Granule production was certified to the standard certification of the TCM National Drug Regulatory Authority (41).
Interventions modifications
Modification or discontinuation of the intervention will be decided by the PIs in each center, according to the requests from participants, or when AECOPD occurs which indicates the need for ICU admission, or when unexpected adverse effects happen.
Compliance
MBYF are free as study drugs and will contribute to subjects bimonthly. All unused packs of drugs and empty bags will be returned to investigator bimonthly. Compliance will be calculated by counting drugs or empty bags: Compliance % of medication = [actual dose/ (specified daily dose × days)] × 100%. Total medication consistency ranging from 80 to 120% will be eligible for the protocol analysis set. Parts of the laboratory tests will be performed on standard schedule freely, which aids in the monitoring of adherence.
Concomitant care
Any drugs or care are permitted when AECOPD occurs. Other ICS and bronchodilators are prohibited during a stable period. Herbal drugs not related to “Tonifying Shen” are allowed to use as disease needs.
Data collection
Items to be measured and the time window of data collection are shown in Table 4. After screening and completing baseline evaluations, participants will join in a Wechat group where we will provide free consultation by physicians. The participants and their family member will be informed that standardized treatment is beneficial to reduce COPD exacerbation, which will reduce medical expenses the benefits. Telephone calls will be conducted on communication monthly.
Data management and monitoring
All the information in the CRF table will be recorded in a specialized clinical experimental database. Details of data management procedures are similar to the other protocol which we have published(42)(https://www.ncbi.nlm.nih.gov/pubmed/32883322).
The data monitoring committee is composed of an investigator from Huashan hospital due to no sponsor. A data monitoring committee will audit the trial conduct via internet meeting monthly and site investigation semi-annually. An interim analysis will be conducted to evaluate the safety of MBYF when 30 subjects have been followed up at 180-days. PIs and the ethics committee have access to these results and will make the final decision to terminate the trial. For all adverse events, the time, duration, treatment measures and outcomes, the severity of the disease, and the association with the study drug will be evaluated and recorded. If an AE occurs, the doctor will immediately take emergency measures and report it to the PIs and the ethics committee within 24 h.
Background information
Background information includes demographic data(gender, age, height, weight and so on) and general clinical data(medical history, course of the disease, treatment history, combined diseases, concomitant medications and so on), which will be recorded during the 1-week screening period. The participants’ information and privacy will be strictly protected and forbidden to the public.
Safety outcomes
Safety will be assessed at 180 days and 360 days after randomization by electrocardiogram (ECG), chest X-ray or computed tomography (CT), laboratory examinations(blood and urine routine, liver and kidney function, glucose.) and adverse events(AEs). AEs will be recorded all the time during the treatment and observed until disappear.
Primary outcomes
(1)The frequency、time and severity of AECOPD
The frequency、time and severity of AECOPD will be collected at every 60 days after randomization. AECOPD is defined as the patient presents with two of the following symptoms (at least one primary symptom) which last at least 2 days. Primary symptoms: Dyspnea, profuse sputum, purulent sputum; secondary symptoms: Wheezing, pain in the throat, cough and the common cold, etc. The interval of AECOPD between two episodes is defined as at least a week. Time of AECOPD is defined as the duration from acute exacerbation to the apparent improvement of the symptoms (the patient’s self-sensation). The severity of AECOPD is defined as three degrees. Mild: no need for clinic service; oral intake of hormones, antibiotics and oxygen therapy are necessary. Moderate: clinic service is necessary, and oral intake of hormones, antibiotics or oxygen therapy is required. Severe: hospitalization or emergency treatment is needed.
(2) COPD assessment test (CAT) score
CAT score will be used for evaluation of symptom at baseline、180 days and 360 days after randomization. Please see the supplementary file.
(3) Pulmonary function tests(PFTs)
A post-bronchodilator forced expiratory volume in 1 second (FEV1 ) % predicted 、forced vital capacity (FVC) ratio (FEV1 /FVC) and blood gas analysis will be measured at baseline、360 days after randomization. Before the test, the treatment drugs should be discontinued for more than 6 hours, violent activity should be discontinued for 2 hours, and patients should rest for 15 minutes.
2.7.4 Secondary outcomes
(1) Modified Medical Research Council (mMRC) dyspnea scale
mMRC will be used for evaluation of dyspnea at baseline、180 days and 360 days after randomization. Please see the supplementary file.
(2) Six minutes walking test (6MWT)
6MWT will be used for evaluation of aerobic exercise capacity at baseline、180 days and 360 days after randomization. The patient will be asked to walk quickly for 6 minutes and the distance will be recorded. Dyspnea scales, fatigue, respiratory rate, heart rate and oxygen saturation before and after walking are calculated. Borg scale will be used to evaluate the severity of dyspnea and fatigue. Please see the supplementary file.
(3) BODE index
BODE will be used for prediction of the mortality rate at baseline、180 days and 360 days after randomization. It includes the following aspects: ①B: body mass index (BMI) ②D:Degree of airflow obstruction ③E:Evaluation of dyspnea ④D:Dynamic ability. The four scores were added together to get the BODE score. Please see the supplementary file.
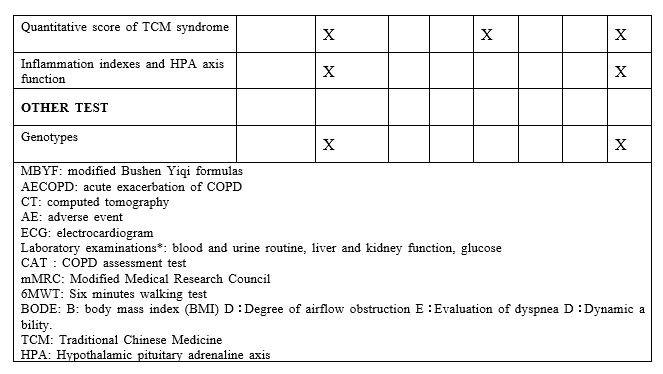
(4) Quantitative scores of TCM syndrome
Quantitative scores of TCM syndrome will be used for evaluation of the TCM syndrome of Fei_Shen_Qi_Xu at baseline、180 days and 360 days after randomization. The treatment index (n)= scores before the treatment-scores after the treatment)/scores before the treatment 100%. Please see the supplementary file.
(5) Inflammation indexes and hypothalamic pituitary adrenaline(HPA) axis function
Inflammation indexes and HPA axis function will be evaluated at baseline、180 days and 360 days after randomization. Draw 10 ml of blood and separate the serum (2-3ml) which is sent to Huashan Hospital for detection of TNF-α, etc. Draw 5 ml of whole blood and separate the blood plasma(1-2ml)to detect hydrocortisone.
Other tests
5 ml Blood will be drawn at baseline, and peripheral blood mononuclear cells will be isolated to detect CRHR1, FCER2, GLCCI1 and other genotypes. The relationship between drug efficacy and genotype will be evaluated.
Sample size calculation
According to the primary outcomes in the previous trial(14, 19), at the 5% significance level, a total of 118 patients per group will be required for a 2-group, the 1-sided calculation to achieve 95% power and the differences of (10.30±6.31, 12.95±5.99) in CAT mean score between the TCM treatment group and placebo group, as calculated using G-power 3.1 software. A loss of 15-20% to follow-up is predicted based on experience——this increase the sample size to 142 participants per group. Based on a 1:1 ratio, the final decision is 300 in total.
Adverse events(AEs)
AEs are defined as negative or unintended clinical manifestations following the treatment. Patients will be asked to report to the investigators any abnormal reactions occurring at any time during the trial. Besides, investigators will collect information about abnormal reactions every 60 days. All details include the time of occurrence, degree and duration of AEs, suspected causes and the effective measures and outcomes will be recorded on CRFs. Subjective discomfort, abnormalities of ECG, chest X-ray or CT and laboratory, should be taken seriously. Careful analysis and immediate measures will be taken to protect the safety of the subjects until the AEs disappeared. There is also a data safety monitoring board to oversee the trial.
Quality control of data
CRFs will be used for data collection. All investigators will receive pretrial training on patients screening, data filling, medication use, AEs reporting and other matters. A trial inspector will visit each hospital regularly to check the electronic database and ensure the protocol is strictly followed. The PIs will be in charge of data validation. and take measures to control the drop-out rate within 15%.
Planned analysis
Baseline data and efficacy will be analyzed using the modified intention-to-treat (MITT) population, and the safety outcomes were analyzed using the safety analysis population (SAP). The Kolmogorov-Smirnov test will be used to determine whether or not continuous variables followed a normal distribution. Unnormal distributional variables will be log-transformed to approximately normal distributions before analysis. Continuous variables will be expressed by Mean±SD or the Median, categorical variables will be shown as counts and percentages. Differences among the two groups will be compared with the use of analysis of variance(ANOVA)for continuous variables and the X2 test for categorical variables. Fisher’s least significant difference test will be used to explore further and compare the mean of two groups. The analysis of covariance will be used for controlling potential confounding variables. Statistical package for social sciences for windows version 16.0 (SPSS, Chicago, IL, USA) will be used for analysis. Two-sided tests will be used throughout, with the statistical significance level set at 0.05, and 95% CIs will be used for interval estimation. The methods of analysis of each variable are summarized in Table 5.
Ethics and dissemination
This study has been approved by the Ethics Review Committee of Huashan Hospital affiliated to Fudan University, Shanghai, China (Provisional Trial No. (425) 2018), and registered at the Chinese Clinical Trial Registry http://www.chictr.org.cn/ at15 December 2019(ChiCTR1900026124). Written informed consent will be acquired from all patients before treatment.
Patient and public involvement
No patient or public was involved in the design of this study. The results will be disseminated to the public through academic conferences and peer-reviewed journals, and briefly sent to all participants through Wechat.
{kind=link}
{kind=link}