High-quality assemblies of thirteen representative wild rice species
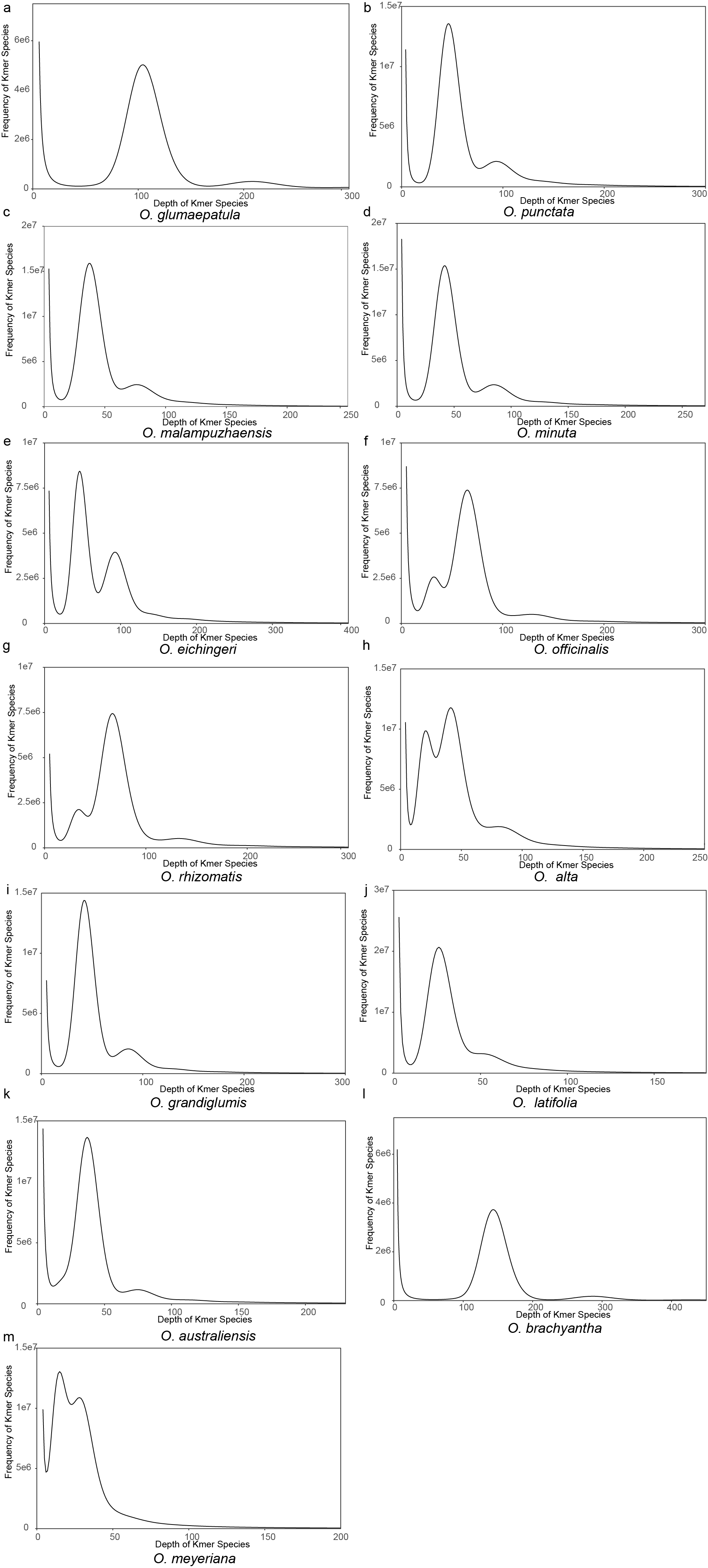
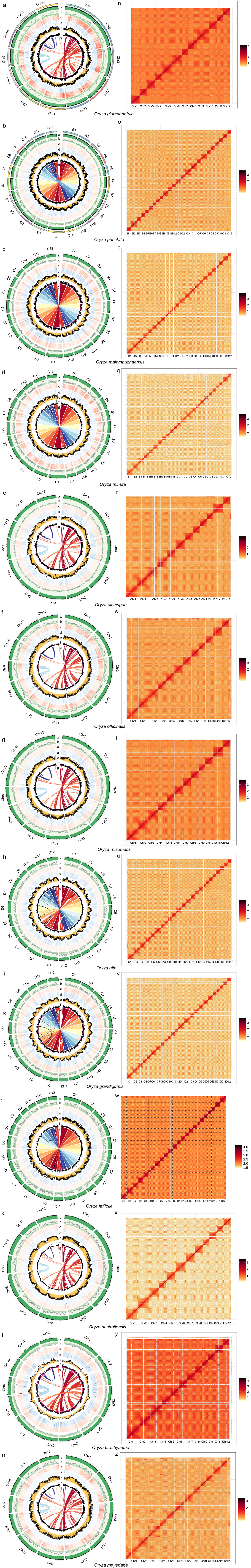
We selected thirteen representative wild rice species, including six allotetraploids and seven diploids, for de novo genome assembly. These accessions exhibit significant variation in geographical distribution and phenotype, as illustrated in Fig. 1a. A total of 331.3 Gb of High-fidelity (HiFi) reads were generated for the 13 wild rice accessions, representing approximately 40-fold coverage relative to the NIP genome size of around 400 Mb (Supplementary Table 1). Through the integration of high-throughput chromosome conformation capture17,18, all 13 wild rice accessions were assembled at the chromosome level, resulting in genome size ranging from 393.77 Mb to 903.31 Mb with an average N50 contig size of 36.80 Mb (Supplementary Table 2, Supplementary Fig. 1, Extended data Figs. 1, 2). The mapping of Illumina short reads and HiFi reads to the 13 wild rice genome assemblies revealed high percentages of alignment, with the results of more than 99.88% and 99.95%, respectively (Supplementary Fig. 2). While an average of 99.44% of the 1614 single-copy orthologs (BUSCO) were identified in these assemblies19. Furthermore, an average value of 23.64 for the LTR assembly index (LAI) was calculated for all assemblies20, as along with high consensus quality values, indicative of their superior quality, continuity and completeness (Supplementary Table 2, Supplementary Fig. 1).
The gene structure annotation was performed using a combination of de novo, homologous, and transcript prediction methods for gene structure annotation based on the repeat-masked genome. This resulted in a higher number of protein-coding gene numbers (ranging from 37,711 to 85,846) compared to previous report on O. brachyantha and NIP21 (41,096) (Supplementary Table 2). The transcript data supported a high percentage (ranging from 71.1–87.7%) of the predicted protein-coding genes, indicating the quality of the gene annotations (Supplementary Table 1). A phylogenetic tree based on 3555 single-copy orthologous genes grouped the 17 rice species into 5 clusters, which contradicted previous findings22 (Fig. 1b). Phylogenetic inference using 1000 randomly selected single-copy orthologs showed that 66.42% were supported, suggesting potential incomplete lineage sorting or gene flow (Fig. 1b). Additionally, an ML tree using chloroplast data was constructed to trace the maternal progenitors of allopolyploid wild rice (Supplementary Fig. 3). Transposable elements (TEs) play a significant role in shaping large plant genomes and driving genome evolution through periodic bursts of amplification (Supplementary Fig. 4). The TE content in the 13 wild rice genomes ranged from 35.11–76.35%, with long terminal repeat retrotransposons (LTR-RTs) being the most abundant (Fig. 1).
Construction of the super pangenome of the Oryza genus helps to review untapped genes and hidden genomic variations
The rice pangenome was expanded to include 16 species (17 subspecies) in the Oryza genus, incorporating genomes from three AA-genome Oryza species. Through OrthoFinder analysis23, a super pangenome was constructed, clustering 808,478 predicted gene models from 13 wild rice species, along with three previously published AA genotype rice genomes and a reference genome of Oryza sativa (Nip), resulting in a pangenome cluster of 101,723 (Extended data Fig. 3a, Extended data Fig. 4a). A total of 9.66% of the gene families (9,834) were found to be shared among all 17 rice accessions, classified as core gene families. Dispensable gene families, present in 2–15 individuals, made up 56.84% of the Oryza genus pan genome, while 33.48% were identified as species-specific gene families (Extended data Fig. 3a, b). Compared with cultivated rice, our super pan-genome of this study can provide an additional 63,881 new gene families.
The 17 rice accessions, comprising 11 diploids and 6 allotetraploids, were categorized into diploid genome types labeled A to G (Extended data Fig. 4b). Additionally, a syntenic pangenome was constructed to analyze the differentiation among the 7 diploid genomes, revealing 16,849 core gene families shared across these genomes, Dispensable families, accounted for 29.73% of the total gene sets (Extended data Fig. 4, Supplementary Table 3), with the largest proportion represented by genome type private gene sets, unique to individual genome types, making up 52.90% of the total gene sets (Supplementary Table 3).
Within the Oryza genus, approximately 81.14% of the core genes could be assigned to protein domains in our Pfam and InterPro databases, which is nearly twice as high as the percentage of dispensable genes (41.82%) and more than seven times higher than accession-specific genes (10.83%) (Extended data Fig. 3d). Core genes exhibited 6- to 20-fold higher expression levels compared to shell and private genes (Extended data Fig. 3e). This tendency was also reflected in gene length, with core genes showing significantly lower (0.15-fold on average) pairwise nonsynonymous substitution/synonymous substitution ratios (Ka/Ks) compared to the dispensable genes (Extended data Figs. 3f, g), indicating conservation of function among core genes in the Oryza genus, while variable genes evolved more rapidly to adapt to diverse environments. The average LTR insertion ratio of core genes was notably lower than that of shell- and species-specific genes (Extended data Fig. 3i), likely due to the lower number of exons per gene in accession specific genes compared to dispensable and core genes (Supplementary Fig. 5), suggesting that exon shuffling or loss contributes to their specificity of genes to each species.
Intriguingly, the core genes in the Oryza genus are primarily involved in fundamental functions such as transposition, iron ion binding, transport, and electron transport, indicating their role in maintaining essential activities of Oryza genus (Extended data Fig. 3h, Supplementary Table 3). A further GO analysis of the different genome types of rice private genes showed distinct functional difference except for disease resistance (Supplementary Fig. 6). These Oryza genus super pangenome open the door for non-AA genome wild rice resources utilizing in rice biology and breeding.
Transposon signature contribute to various genome and centromere size in rice
The selective removal and retention of TEs has significantly influenced the genome size, adaptation and evolution of the Oryza genus 24. Scientists have been intrigued by the question of which specific subfamily of TEs influences the genome size25. To address this, a comprehensive classification of TEs within the Oryza genus was conducted, along with an in-depth analysis of the expansion profiles of long terminal repeat (LTR) subfamilies (Top 6 selected from the largest genome size in Oryza) were performed. Each LTR-RT sub-family displayed unique patterns of amplification across the species, impacting gene numbers and expressions (Extended data Fig. 5b, Supplementary Fig. 7a, Supplementary Table 4). In addition to the amplification of Gypsy superfamily (Ogre, Retand, and Tekay), the Angle LTR belongs to the Copia superfamily emerged as a significant contributor to the large genome size of O. australiensis (E genome type rice), distinguishing it from other rice species (Extended data Fig. 5b, c, Supplementary Fig. 7a). The genome sizes of B, C, and D type rice genome were primarily influenced by the top three Gypsy superfamilies in descending order: Ogre, Retand and Tekay (Extended data Fig. 5c, Supplementary Table 4). The genome size of G genome type (O. meyeriana) was predominantly influenced by Retand LTR amplification (Extended data Fig. 5c), and the abundance of Tekay in O. glumaepatula surpassed that in the O. sativa resulted in its slightly puffy genome. Notably, O. brachyantha (F genome type) exhibited significant elimination of all LTR subfamilies compared to O. sativa (Extended data Fig. 5c), aligning with the observed LTR density patterns across Oryza genomes (Supplementary Fig. 8). Genome size showed a stronger correlation with the Retand LTR superfamily compared to the other subfamilies within the Oryza (Extended data Fig. 5d). While SIRE and the CRM subfamily made up a small portion of the entire genome, they influenced a certain proportion of genes expression (Extended data Fig. 5c, Supplementary Fig. 7b, Supplementary Table 4).
The distribution of whole-genome intact long terminal repeats (LTRs) indicated that the majority of LTR bursts occurred in proximity to centromeric regions (Supplementary Fig. 8). Rice centromeres consist of organized satellite repeats (SRs), interrupted by centromere-specific retrotransposons (CRRs) 26. It remains unclear if other lines or wild rice plants possess unique centromere satellites and whether centromere repositioning events occurred during centromere evolution. In cultivated rice (MH63 and ZS97), 155 bp and 165 bp CentO satellite repeats were categorized into seven distinct subsets across the 12 chromosomes 27. Interestingly, only a few copies of satellite repeats were identified in O. meyeriana and O. branchyantha wild rice. Compared to the cultivated rice (NIP) genome, the C genome contains centromere-specific 126 bp and 366 bp Cent satellite repeats (Supplementary Table 4). Phylogenetic analysis results showed that the satellite repeats in Oryza can be classified into four groups (Extended data Fig. 5e), with chromosomes of the same type tending to cluster together, supporting models of repeated amplification events involving the central domain and local homogenization. Examination of the genetic characteristics of centromeres in the Oryza genus revealed a decrease in gene density as centromeres are approached, along with an increase in transposon density and the frequency of k-mers (Extended data Fig. 5f). While the wild rice centromere sizes, except Oryza brathyantha, were notably larger than those in cultivated rice, the opposite trend was observed for the number of genes (Extended data Fig. 5g, Supplementary Fig. 7c). A comparative sequence map of centromere synteny between cultivated rice and wild rice highlighted extensive structural rearrangements in centromeric and pericentric regions across the Oryza genus (Extended data Fig. 5h, Supplementary Fig. 9, Supplementary Table 4). Additionally, several centromere repositioning events were noted in the synteny analysis (Supplementary Fig. 9).
Large-scale chromosomal rearrangement and inferring the genome evolutionary history of Oryza lineages
To compare the karyotype stability between cultivated rice and wild diploid genomes, we created a synteny map and conducted whole-genome pairwise alignments to identify large segment variations, such as translocations and inversions7. Notably large inversions (with more than 5 consecutive genes) shared by at least two consecutive species in the Oryza genus were prevalent in the genome alignments11 (Extended data Fig. 6). While reports on segregating inversions in wild rice are scarce and have not yet included natural polyploid wild rice, most of these events were observed in the low-recombining pericentromeric regions of the Oryza chromosomes, with a few inversions being species-specific (Extended data Fig. 6). The AA genomic type genome displayed a significant level of chromosomal conservation. Furthermore, the gene synteny findings provide support for the phylogenetic position of the rice genus (Supplementary Fig. 10). Through multiple species/genome comparisons, many large-scale genomic rearrangements were validated, including an inversion of an approximately 2.53 Mb segment comprising 166 genes on Chr6 in the modern cultivated rice NIP (Fig. 2a). The inversion occurred only among the common wild rice and Oryza sativa japonica with high latitude distribution, indicating that it has risen to expansion of geographical range. OsMFT1, which contribute to its later flowering in Oryza sativa was located in the inversion region with an 89 kb distance next to the breakpoint in Oryza glumaepatula 28(Fig. 2b).
Chromosomal rearrangements involving homoeologous groups 1, 3, and 6 of allotetraploid wild rice were initially identified in Oryza species (Extended data Fig. 7a). The comparison of syntenic blocks revealed that chromosomes 6Dt in O. latifolia displayed complete collinearity with the corresponding 3Dt chromosomes in O. alta and O.grandigumis, However, a reciprocal translocation was observed between 3Dt and 1Ct in O. alta and O.grandigumis (Extended data Fig. 7a), due to a fragmental collinearity between 3Ct in O. latifolia and 1Ct in O. alta and O.grandigumis. Additionally, a translocation between 1Ct and 6Ct as identified, with the 1Ct segment translocated to the end of 6Ct. Furthermore, a translocation was detected in homoeologous groups 4 and 7 in Oryza (Extended data Fig. 7c-i).
By aligning allotetraploid wild rice resequencing data to the corresponding diploid BB and CC or CC and EE genomes, homeologous exchanges on each chromosome were identified based on the coverage depth calculated from unique reads. Several translocations of large segments between the subgenomes post-tetraploidization were discovered (Extended data Fig. 7b, c). Chromosomes Bt1 and Ct1 exhibited high synteny, but the coverage depth of the reads to the BB and CC genomes indicated a significant homoeologous exchange between them (Extended data Fig. 7b). The potential history of homeologous exchange is depicted in Extended data Fig. 7d.
Discovery of untapped SVs in the Oryza genus sequence and their influence on agronomic traits
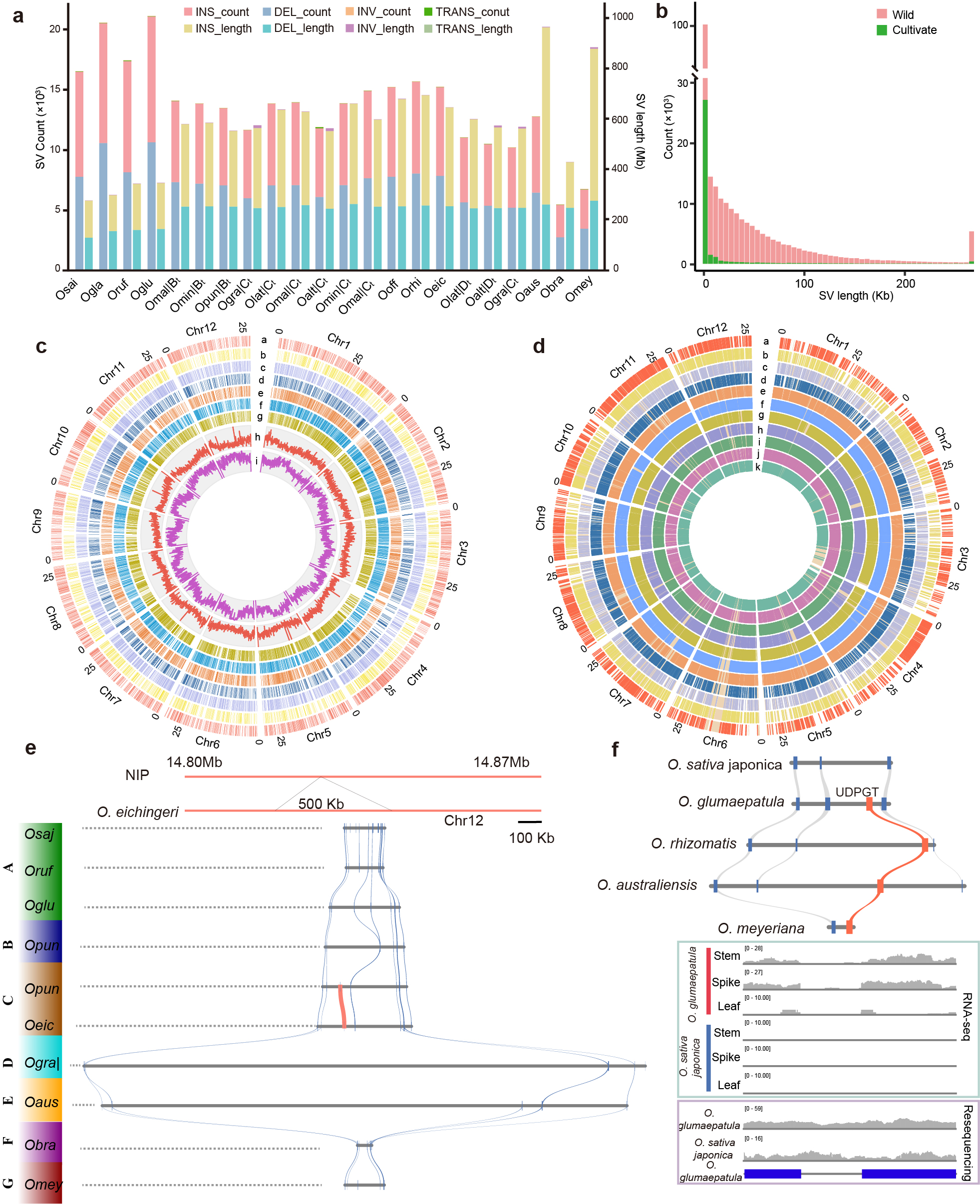
Despite extensive efforts to analyze genetic variants in cultivated rice and its ancestor species O. rufipogon 29, the genetic diversity in distantly related wild rice species such as O. punctata, O. rhizomatis, and O. meyeriana remains poorly understood. We identified 2781–10656 insertions, 2680–10419 deletions, 4–52 translocations, and 7–22 inversions in the 16 rice accessions, with sizes ranging from 162.49-278.65 Mb, 182.13-705.17 Mb, 8.64-887.29 kb, and 41.51–11.33 Mb, respectively (Extended data Fig. 8a, Supplementary Table 5). Interestingly, the cultivated rice and the AA genome of wild rice showed a higher number of structural variations compared to the non-AA genome of wild rice, although the size of variation was smaller, likely due to more regions aligning with the reference genome (Extended data Fig. 8a). Wild rice species-specific SVs accounted for a significant portion of the total variation, indicating untapped genetic diversity in wild rice compared to cultivated varieties (Extended data Fig. 8a).
The majority of insertions, deletions, and inversions in the cultivar were shorter than 5 kb. As the length of SVs increased, there was a significant decrease in the number of SVs in cultivar and wild rice, whereas the wild rice variety had a higher number of SVs larger than 250 kb, leading to the presence of numerous private genes in the wild rice genome (Extended data Fig. 8b, Extended data Fig. 9a). Intergenic regions within the Oryza genus were most common locations for SVs, followed by regions ± 10kb around genes (Extended data Fig. 9b, Supplementary Table 5), in line with previous research findings 9. Surprisingly, insertion and deletion variations were less frequent at the chromosomes ends (Extended data Fig. 8c). The private genes in each rice genome type and their corresponding presence-absence variations (PAVs) relative to the NIP were also recorded (Extended data Fig. 8c, d). when using NIP genome sequence as a reference, the cultivated rice displayed a higher number of SVs compared to wild rice, consistent with earlier observations (Extended data Fig. 8a). However, wild rice, particularly O. australiensis and O. meyeriana, exhibited substantial variation in SV sizes, indicating an enrichment of SVs in repetitive DNA regions (Extended data Fig. 9c). Further examination of transposable elements in PAV sequences revealed that other and DNA transposable elements were the primary components of both deletion and inversion variation (Extended data Fig. 9d).
By analyzing a large number of SVs across different rice genomes within a phylogenetic framework, we were able to uncover evolutionary events that would have otherwise gone undetected with a limited number of genomes. Recent findings suggested that gene loss could be linked to insertion/deletion event. For example, a 500 kb insertion corresponding to the NIP genome was identified on chromosome 12 at 14.50 Mb (Extended data Fig. 8e). In addition, the insertion occurred only in the O. eichingeri and Ct subgenome of O. punctata. Further detailed investigation revealed that the PAV region contained a gene (OPUW363G084108/OEIW71G043491) specific to the C genome wild rice30 (Extended data Fig. 8e). This gene might be de novo birth to contribute to the ability of wild rice to adapt to poor and problem soil in Sri Lanka. The phylogenetic results revealed that the gene originated from O. eichingeri and then transferred to O. punctata, providing evidence that O. eichingeri was the progenitor of tetraploid O. punctata, consistent with our chloroplast evolution results. This result demonstrated that insertion variation occurred during C genome wild rice speciation and cultivated rice, which exhibited SVs possibly via introgression from hybridization with O. eichingeri.
A multitude QTLs in O. officinalis have been identified for brown planthopper resistance, but the lack of unknown sequences in wild rice has hindered the cloning of these genes31. In this study, we identified the Bph4 gene through a combination of comparative genome analysis and gene annotation within the QTL region (Supplementary Fig. 11). Haplotype analysis indicated that Bph4 is highly conserved in cultivated rice but displayed diversity in wild rice.
Among the 13 wild rice accessions studies, only three species retained the functional S28 locus32, while others lacked either the ribosomal protein S27 gene or the nearby UDPGT gene (Extended data Fig. 8f). The phylogenetic analysis of the Oryza genus suggested that the HS locus likely originated from O. australiensis and diverged from the C genome of wild rice (Extended data Fig. 8f).
Allelic and regulatory elements variations
The natural allelic variation of genes is essential for phenotypic diversity, environmental adaptation, and the process of domestication 33–35. Our analysis focused on variations in whole-genome alleles and their regulatory sequences (gene ± 10kb) in the rice genome, as there are very few highly collinear blocks between non-AA genomic wild rice and cultivated rice (Extended data Fig. 10, Supplementary Table 6). As the divergence from cultivated rice increased, the number of colinear genes between wild rice diploids and cultivar rice decreased, ranging from 18,463 to 23,812, with an average of 20,288 (Supplementary Table 6). Including 19 published, high-quality, chromosome-level cultivated rice genomes (Supplementary Table 1) in our study allowed us to identify comprehensive SVs resources for both wild and cultivated rice9. By mapping collinear genes with 10 kb nearby regions onto the corresponding region of Nipponbare, we identified SNPs and InDels of 50 bp or greater as PAV targets. The total number of SVs increased with the accession number, with cultivated rice showing a higher percentage of nonredundant SVs compared to wild rice (Extended data Fig. 11a). The wild rice genomes exhibited a greater number of alleles and gene haplotypes than cultivated rice (Extended data Fig. 11b, c), indicating a rich source of novel genetic variations. To delve deeper into the functional impact of SVs on genes or proteins, combining variant alleles detected in each species into haplotypes and annotating each accession independently is essential. Wild rice (sub)genome displayed a higher number of alleles in collinear genes within the core genome compared to cultivated rice (Extended data Fig. 11c). The number of gene haplotypes (gHap) and gene-coding sequence (CDS)-haplotypes (gcHap) in wild rice was significantly greater than in cultivated rice (Extended data Fig. 11c). Analyses of protein diversity in collinear genes between wild and cultivated rice have provided insight into their functional differentiation. A genome-wide protein cluster was created based on their domain similarity, revealing that wild rice had approximately 7 clusters, corresponding to the number of wild rice genome types, whereas cultivated rice predominantly clustered into one group, corresponding to the AA genome type (Extended data Fig. 11d). Furthermore, analysis of gene presence-absence variations (PAVs) distinguished major species and highlighted significant differences between wild and cultivated rice (Extended data Fig. 10b-d). The majority of group-unbalanced genes, accounting for 87.33%, were more prevalent in wild rice but less common in cultivated rice, underscoring the substantial legacy of mutations in wild rice (Extended data Fig. 11e). Notably, the selection for grain coat color during rice domestication is evident, with wild rice species predominantly displaying black and red grain, while most cultivars exhibit white seed coat color. Structural variation analyses revealed distinct haplotypes of the Rc protein among cultivated and wild rice accessions36. Compared to the Rc haplotype in cultivated rice, wild rice exhibited 7 haplotypes corresponding to different genome types, suggesting that genetic divergence in Rc played a role in grain pericarp development during domestication (Extended data Fig. 11f).
Gene CNVs and NLR repertoire in rice
Recent studies have highlighted the significant role of genomic copy number variations (gCNVs) in the evolution and domestication of crops 37,38. However, the accurate identification of gCNVs in highly repetitive genome sequences within the rice genus pose notable challenges. Leveraging our high-quality assemblies, we systematically investigated gCNVs by aligning collinear blocks of the rice accessions against the Nipponbare reference genome, assessing their potential impact on important agronomic traits. Through whole-genome comparisons, we identified 207 genes with tandem repeats across the 14 wild rice assemblies, potentially influencing yield, resistance, grain quality, heading date, biotic and abiotic resistance (Supplementary Table 7). To gain further insights into the functional roles of gCNVs in rice, we analyzed 4400 genes with known functions from a previous study39. Among these genes, 36 exhibited tandem repeats in the rice genus, impacting various agronomic traits related to yield, disease and pest resistance (e.g., blast, bacterial blight, rice brown planthoppers), biotic stress tolerance, element transport, and other important adaptation traits like heading date and hybrid sterility (Fig. 3a). Additionally, we assessed the expression levels of selected gCNVs to investigate potential alterations in their expression profile. Notably, several variations linked to the Pi9 cluster, with gCNVs in the 10.38 Mb region of Nip genome chromosome 6, were also identified. Pi9 is a well-known gene in rice that offers strong and long-lasting resistance to the fungus M. oryzae40. Interestingly, Pi9 is a typically NLR genes with copy number variation, which contributed to rice species environmental adaptation (Supplementary Fig. 12).
Genes that encoding nucleotide-binding domain and leucine-rich repeat (NLR) proteins play a crucial role in plant immune systems 41. Therefore, it is essential to have a comprehensive and accurate NLR dataset for rice genera. Plant NLRs often occur in clusters, making their identification challenging. To address this issue, we utilized RGAugury 42 and DupGen_finder 43 tools, resulting in a total of 7,048 NLR genes across rice genus (Supplementary Table 7). The number of NLR genes varied from 419 in O. glabberima to 511 in O. sativa indica (R498) in cultivated rice and from 159 in O.australiensis to 669 in O. punctata in wild rice (Fig. 3b, Supplementary Table 7), This suggests that the immune system in wild rice has a more diverse evolutionary history compared to cultivated rice.
Our study focused on identifying and categorizing NLRs in different rice species to establish a comprehensive understanding of NLR diversity within rice genus. Interestingly, the diploid rice genome exhibited a lower number of NLR in wild rice compared to cultivated rice, despite the larger genome size in wild rice (Fig. 3f). For instance, the genomes of O. australiensis and O. meyeriana, although twice the size of NIP, contain only half the number of NLRs of that in cultivated rice (Fig. 3f). Analysis of NLR distribution showed that while R gene singletons were similar between wild and cultivated rice, cultivated rice tended to have a higher number of R genes in pairs or clusters compared to wild rice (Fig. 3b-d). Redundancy analysis revealed that 55.64% of NLR signatures were shared across all genomes, with 15 unique signatures in the cultivated group and 162 unique signatures in the wild group (Fig. 3g). The study found that as the number of cultivated rice accessions increased, the number of core NLR signatures also tended to increase. Redundancy analysis of the NLR gene in wild and cultivated rice revealed that 78.8% of the NLR genes in cultivated rice were dispensable, slightly lower than in wild rice (Fig. 3h). More than 90% of NLR genes in the core NLR genome were expressed in both wild and cultivated rice, while around 20% of NLR genes in the dispensable genome exhibited low or no expression under normal conditions, suggesting specific expression upon encountering disease (Fig. 3i).
We classified NLRs of the rice genus into 369 clusters, and 167 clusters of which were sharply increased in cultivated rice (Supplementary Table 7), including well-studied rice R gene families that provide resistance to rice blast disease caused by Xanthomonas oryzae. Pv. Oryzae (Xoo), such as WRKY61 (Fig. 3j). Additionally, an NLR expansion event was observed in the wild rice pangenome (Fig. 3j), enabling these plants to adapt to various environments compared to cultivars. By leveraging lost NLR gene rice during domestication and artificial selection, we can enhance the resistance resources of cultivated rice and enrich the diversity of modern commercial rice. The total number of NLRs in cultivated rice species has increased compared to diploid wild rice species, despite some NLR gene losses, indicating that NLR expansion into cluster forms may be driven by breeding for specific pathogen resistance.
{kind=link}
{kind=link}
{kind=link}