Our current study demonstrates that HCMV could infect EPCs, resulting in the functional impairments of EPCs. The initiation of p38 MAPK signaling pathway and its subsequent activation of the transcription factor AP−1, induced by HCMV infection, plays a vital role in regulating the secretion of TGF-β1 in BM-EPCs.
Emerging evidence demonstrates that the EPCs abnormality is associated with PGF development post-transplantation [2], but the mechanism of EPCs impairments in PGF patients is not clear. Clinical data indicates that HCMV infection is an important risk factor for PGF [4, 5]. However, no previous study has focued on the impact of CMV on BM-EPCs.
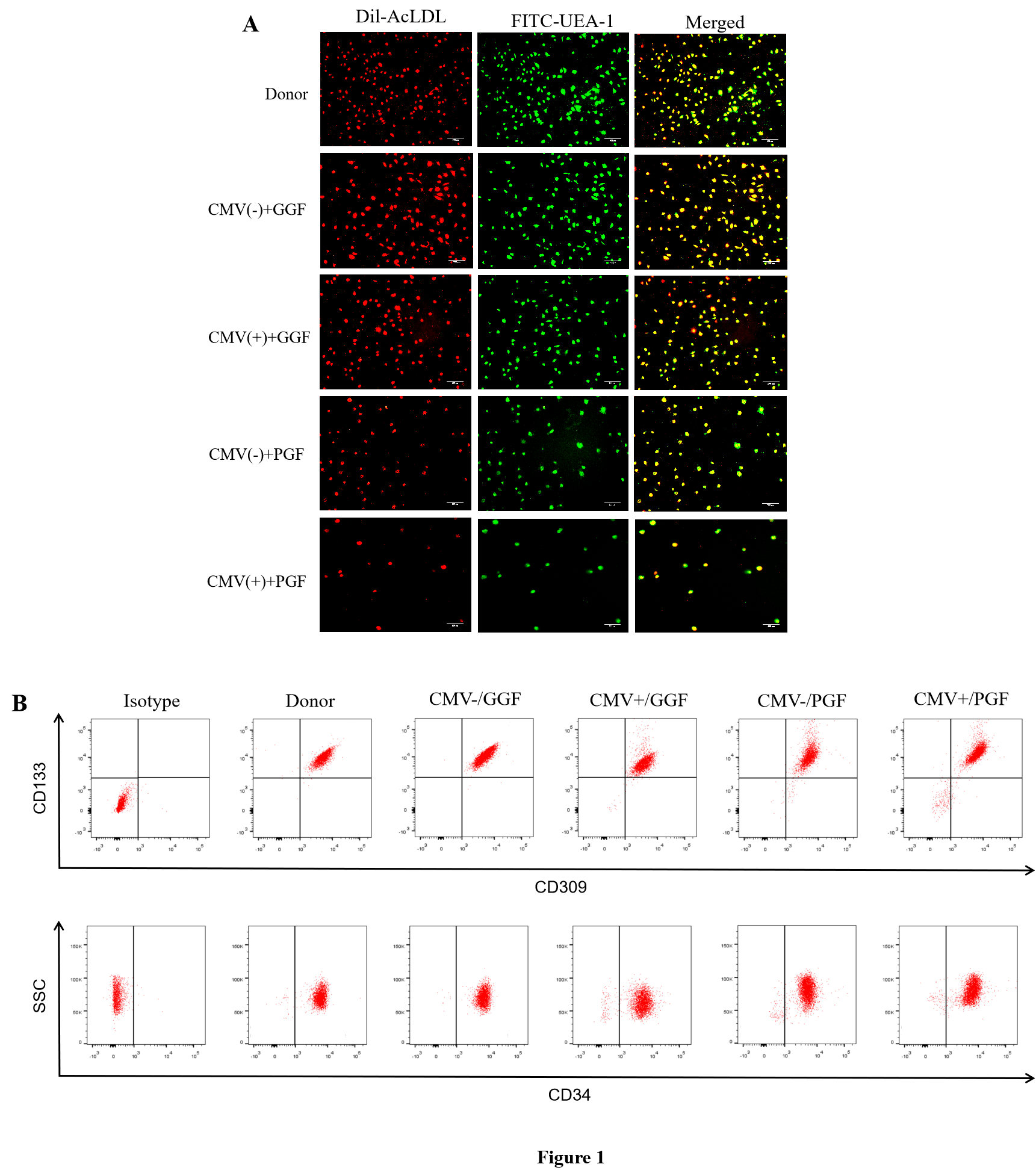
Shi et al. proved the dysfunction of BM-EPCs were found in patients with PGF, which encompased alterations in their proliferative capacity, migratory potential, susceptibility to apoptosis, and ability to promote angiogenesis [2]. Our findings further substantiated the presence of compromised functionality in BM-EPCs following HCMV infection, consistent with the aforementioned dysfunctional BM-EPCs in PGF. Notably, our results showed that the dysfunction observed in BM-EPCs derived from patients with HCMV-emia accompanied by PGF were more pronounced compared to those derived from patients without HCMV-emia accompanied by PGF, indicating the pivotal role of HCMV in the pathogenesis of BM-EPCs injury.
The process of hematopoiesis is precisely regulated through the action of hematopoietic cytokines [20]. Our study revealed that HCMV-infected BM-EPCs exhibited excessive secretion of TGF-β1, a kind of hematopoietic inhibitory factor, which effectively impedes the growth and proliferation of CD34 + hematopoietic progenitor cells (HPCs) [7, 22, 23]. Moreover, we observed that the introduction of a neutralizing antibody against TGF-β1 effectively reversed this suppression of hematopoiesis caused by HCMV infection, demonstrating that the crucial role of TGF-β1 in mediating the suppressive effects on hematopoiesis induced by HCMV.
Shi et al [2] proved that the crucial involvement of p38 MAPK activation in the dysfunction of BM-EPCs observed in patients with PGF, highlighting the pivotal role of MAPKs in the pathogenesis of PGF. AP−1 transcription complex, including fos and jun is one of the most important substrates of MAPK [17]. Previous study demonstrated that AP−1 was highly active following HCMV reactivation and was a determinant of HCMV reactivation [21]. In addition, activation of the AP−1 transcription complex, including fos and jun, contributes to TGF-β1 expression [18, 19, 24]. Therefore, we were specifically interested in the question of whether HCMV caused AP−1-indepent induction of TGF-β1 expression is mediated via the p38 MAPK pathway. Our results showed that the activation of phospho-p38 was not significantly affected by the administration of T−5224 in HCMV-infected BM-EPCs, while addition of SB203580 significantly downregulated the activation of phospho-c-fos, which demonstrated that phospho-c-fos was the downstream target of phospho-p38. Addition of either SB203580 or T−5224 could restores secretion of TGF-β1 and myelopoiesis to levels observed with BM-EPCs in Normal control group. These results demonstrated that HCMV induced increased TGF-β1 secreted by BM-EPCs mainly through MAPK p38-AP−1(c-fos) pathway.
In summary, HCMV could infect BM-EPCs and lead to their dysfunction. Enhanced secretion of TGF-β1 by BM-PECs is induced by HCMV through the up-regulation of p38 MAPK and its downstream transcription factor AP-1, resulting in myelosuppression, which might make a substantial contribution to the pathogenesis of PGF after allo-HSCT, providing innovative therapeutic strategies targeting PGF.
{kind=link}
{kind=link}