5.1. Materials
DCFH-DA (The 3-hydroxytyramine hydrochloride and Diacetyldichlorofluorescein) probe were acquired from Sigma-Aldrich (St Louis, MO, USA). DMEM, penicillin-streptomycin (PS), and phosphate-buffered saline (PBS) were supplied by HyClone, headquartered in Logan, UT, USA. The following reagents and consumables are sourced from the same literature that we have previously published [49]: HaCaT was sourced from Cell Lines Service, Eppelheim, Germany, while high glucose DMEM medium was obtained from Gibco Co., Ltd. Sigma Co., Ltd provided MGO and bFGF, and Beyotime Co., Ltd supplied Cell Counting Kit-8 (CCK-8). C57BL/6J mice (male, 6–8 weeks, 20 ± 5 g) were purchased from Shanghai SLAC Laboratory Animal Co., Ltd., Shanghai, China (SCXK (Hu) 2022–0004). High-fat and standard diets were procured from Shanghai Pu Lu Tong Biological Technology Co., Ltd. Sigma Co., Ltd provided Streptozotocin (STZ). Recombinant Bovine Basic Fibroblast Growth Factor (rb-bFGF, No. 20160102) was purchased from Zhuhai Yisheng Biopharmaceutical Co., Ltd. Antibodies used for immunohistochemistry (IHC) staining included anti-CD31 (diluted 1:2400, CST, #77699), anti-F4/80 (diluted 1:600, CST, #70076), anti-PCNA (diluted 1:1000, Abcam, ab29), anti-NF-kB p50 (diluted 1:36000, Abcam, ab32360), anti-iNOS (diluted 1:100, Abcam, ab283655), anti-CD206 (diluted 1:2000, Proteintech, 60143-1-Ig), anti-Shc (diluted 1:400, Abcam, ab33770), anti-IRS1 (diluted 1:50, Abcam, ab52167), and anti-IRS2 (diluted 1:200, Abcam, ab134101). Bio-Plex Pro Mouse Cytokine IL-1β (171G5002M) and Bio-Plex Pro Mouse Cytokine IL-6 kit (M600003G7V) were purchased from Bio-Rad Laboratories, Inc[49].
5.2. Bacterial culture and biofilm preparation
The standard strains of Gram-positive Staphylococcus aureus (ATCC 29213) and Gram-negative Escherichia coli (ATCC 25922) with good activity were selected for our experiments. The strains were inoculated on the solid agar medium for 12h and then selected a single colony and cultured it in 25mL liquid LB medium at 37 ℃ for 18–24 h. The cultured strains were then harvested by centrifugation and washed with PBS for three times. The concentration of bacterial solution was measured by ultraviolet spectrophotometer at a wavelength of 600nm. According to the colony counting method (CFU), re-adjust the OD 600 value of the bacterial stock solution to 0.1, which corresponds to 1 × 108 CFU/mL bacterial density.
400 µL of the bacterial suspension (1 × 108 CFU/mL) in the LB medium was added to the 24 well plate and cellulose film for the sake of bacterial biofilm. After that, the bacteria were cultured for 2 days, and the culture medium was changed once a day. The culture medium was discarded and washed the unattached bacteria with PBS for three times to harvest biofilms.
5.3. Preparation and characterization of Nisin-PA
The Nisin (10mg, 1mg/mL) was dissolved in 10 mL of ultrapure water under ultrasound (3 min). Afterward, PA (164.7µL, 2mg/mL) was incrementally added to the solution under magnetic stirring, and the reaction proceeded in alkaline solution for 2h. Then the product was dried in vacuo overnight to obtain Nisin-PA.
The products were characterized by UV–Vis absorption spectra, FTIR analysis, and 1H NMR spectroscopy in D2O (Bruker AVANCE III HD 600 MHz) according to the previous literature[58].
5.4. Preparation of NPF
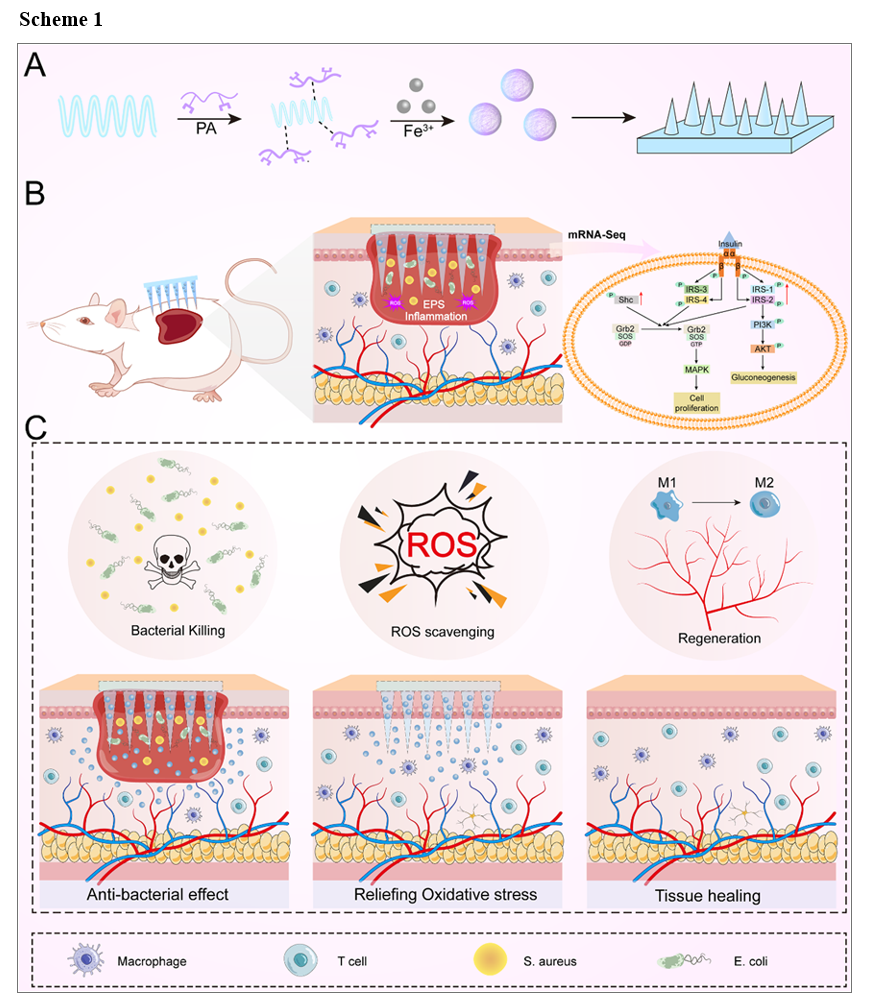
Different mass ratios of PA and Fe3+ (1:1, 2:1, 3:1) were used for constructing a pH-responsive nanocomplex via rapidly assembling of Schiff base and Fe-polyphenol complexation reaction. As an illustrative example, consider the synthesis steps in detail for a 3:1 ratio of PA to Fe3+. Briefly, PA solution (164.7µL, 2mg·mL− 1) was added dropwise to Nisin solution under magnetic stirring for 2h at a speed of 500r. Afterward, FeCl3 (71.4µL, 3mg·mL− 1) was added and continued stirring for 30min to obtain the pH-responsive NPF through Fe-polyphenol complexation interaction. The NPF dispersion was dialyzed in deionized water for 6 hours (MWCO = 8000 Da) and subsequently dried under vacuum overnight to yield the NPF product.
5.5. Characterization of NPF
The particle size distribution, polydispersity index (PDI) and morphology of NPF were measured according to the previous literature methods[58]. The UV-Vis spectrometer was utilized to measure the UV-vis absorption of NPF and the OD value of free Nisin was measured at 270 nm to obtain standard curve. Moreover, the concentration of the NPF was defined by the Fe3+ content analyzed by ICP (ICP-OES, Agilent 720-ES).
5.6. Fabrication and characterization of MN
Hyaluronic acid powder (8000–10000 Da) was dissolved in an NPF aqueous solution to obtain a 30% mass fraction HA solution. For HA MN preparation, the HA solution (w/t 30%) was poured onto PDMS molds under vacuum (∼ 0.08 MPa), dried in a sealed desiccator overnight with the molds, peeled off, and stored in a sealed desiccator at room temperature. The dried NPF@MN were then examined for morphology using a microscope (Nikon ECLIPSE E 100, Nikon Corporation, Japan).
5.7. Insertion capability of the MN
The frozen section test assessed the insertion capability. MNs were applied to isolated rat skin for 15 minutes, followed by embedding the treated skin in OCT compound and freezing it at − 80°C. Subsequently, 10 µm sections were obtained from the frozen sample using a cryostat microtome (CRYOSTAR-NX50, Thermo, USA).
5.8. Hygroscopicity Test
Store the dried MN in a sealed container under 75% humidity. Then MN was taken out and weighed every minute, a confocal microscope was used to take pictures of the microneedle changes in morphology.
5.9. In vitro stability and acid response-ability of NPF
The stability of NPF was detected according to the method in the references[58], and the acid responsiveness of NPF was detected by measuring the size of the sample and observing the morphological changes by TEM.
5.10. pH triggered Nisin release properties
To test and verify the pH sensitive character of NPF at different pH conditions, the release of Nisin from NPF was studied. Briefly, 0.2mL of NPF was well dispersed into 25 mL PBS with different pH (pH = 5.0, pH = 7.4 respectively). Then the suspensions were transferred into dialysis bags (MWCO 8K Da) and stirred at 37°C with gentle agitation (100 rpm/min). Retrieve 2.0 mL of the sample solution at specified time intervals and replenish with an equivalent volume of PBS. The absorbance value of release free Nisin was measured by UV–vis spectrophotometer and analyzed according to the standard curve of the concentration of Nisin.
5.11. In Vitro Antioxidant Tests
The antioxidant performance of samples with different PA-Fe mass ratios (1:1, 2:1, 3:1, 4:1) and different concentrations of NPF (31.25µg/mL-1000µg/mL) were evaluated by measuring the DPPH radical scavenging capacity. Briefly, 1mL of nanoparticle aqueous solution and DPPH ethanol solution (1mL 0.1mM) were thoroughly mixed and stirred at room temperature in the dark for 30min, and then the wavelength of DPPH was scanned with an UV-Vis at a wavelength of 517 nm. The absorbance of the supernatant was read, measured three times parallel, and the inhibition ratio was calculated according to the following formula.
DPPH scavenging (%) = (1-Asample/Acontrol) × 100%
Where A are the absorbance value of the sample at 517nm. The antioxidant performance of the sample under different pH was also tested.
5.12. Detection of Reactive Oxygen Species
The scavenging capacity of NPF against intracellular ROS was detected by DCFH-DA probe, fluorescence imaging was performed by living cell workstation, flow cytometry was used for quantification. First, human fibroblast 3T3 cells were seeded in six-well plates (12×104 cells/mL) and incubated with NPF and NPF@MN for 6 h, the medium was then removed and treated with 0.3mM H2O2 for 30 min. Finally, the cells were incubated with DCFH-DA (10µM) in the dark at 37 ℃ for 20 min. Cell fluorescence intensity was imaged by living cell workstation and quantification of intracellular ROS level by flow cytometry analysis.
5.13. Scavenging of HO• and O2•−
In addition, we also selected two physiological related ROS, hydroxyl radical (• OH) and superoxide anion radical (O2 •−) to test the ROS scavenging capacity of NPF. First, we evaluated the scavenging efficiency of NPF for hydroxyl radicals by measuring the presence of fluorescent 2-hydroxyterephthalic acid. Briefly, a series of NPF (31.25 µg/mL-1000 µg/mL), TA (2mM), H2O2 (40 mM) were prepared in PBS (25 mM, pH = 7.4) solution, and the mixture was incubated overnight on a shaker at 37 ℃ overnight. TA can react with hydrogen peroxide to generate 2-hydroxyterephthalic acid. The clearance rate of NPF to hydroxyl radicals was evaluated by measuring the fluorescence absorption value at 425nm.
The scavenging capacity of NPF against superoxide anion was determined by the following methods. First, a series of NPF (62.5 to 1000 µg/mL) were prepared, and then were added to the solution containing 200µL Riboflavin (20 µM)、200µL methionine (12.5 mM) and 200 µL NBT(75 µM) and were incubated under ultraviolet radiation for 30 min, finally the absorbance of the mixture was measured. The positive control group was the sample containing riboflavin, methionine and NBT. All experiments were conducted in darkness.
5.14. Phenotype Detection of RAW264.7 Cells
After incubating RAW264.7 cells with DMEM and NPF@MN for 24 hours, the treated cells were collected. Subsequently, they were sequentially stained with the CD80/CD206 antibody for 1 hour, and the fluorescence signal of CD80+/CD206 cells was measured using a flow cytometer.
5.15. Planktonic Antibacterial Studies
The antimicrobial properties of NPF@MN against S. aureus and E. coli. was determined through bacteriostatic zone test, time dependent germicidal test, concentration dependent germicidal test and live/dead test.
The MN and NPF@MN were dissolved in PBS solution, and the sterilized round filter paper with a diameter of 10mm was soaked in it. The S. aureus and E. coli (diluted to 106 CFU/mL in LB medium) suspension (100 µL) were inoculated onto LB agar plates. The treated filter paper sheets were then placed on bacteria-coated agar plates, and the inhibition circle formed around the filter paper sheets were observed after incubation at 37°C for 24 h.
For the time-dependent sterilization test, MN and NPF@MN were placed in 24-well plates with 1 mL of PBS at pH 5.5 and 7.4, respectively. 35µL of bacterial suspension is added, and after overnight incubation at 37°C, serial dilutions are transferred to LB agar plates, and colonies are photographed and counted.
The antibacterial activity of various concentrations of NPF@MN was studied by standard plate count method, and the NPF@MN was dissolved in PBS solution and then was added to the 106 CFU/mL E. coli and S. aureus suspension. A 100 µL aliquot of the diluted bacterial solution was applied to agar plates, and colonies were observed.
Live/dead bacteria staining was used to study the viability of bacteria after exposure to MN, NPF and NPF@MN under different pH conditions. Qualitative assays were performed according to the manufacturer method. specifically, bacteria were mixed with dye solutions of SYTO 9 and propidium iodide for 0.5 h at room temperature, followed by imaging with Live-cell workstations. Living bacterial cells fluoresce green, and dead bacterial cells fluoresce red.
5.16. Biofilm Antibacterial Studies
The inhibition ability of NPF@MN on biofilm was determined by crystal violet staining, and the bacterial suspension was incubated in a 24-well plate for 2 days to obtain biofilm. The biofilm was incubated with MN, NPF and NPF@MN (concentration of 300 µg/mL) for 24 h, and then 500 µL of 0.1% v/v crystal violet ethanol solution was added and soaked for 20 minutes. Bacterial growth was studied by measuring the absorbance of crystal violet solution at 590 nm by microplate reader.
The antibiofilm activity of NPF@MN was assessed by the number of CFUs. Inoculate 10 µL of diluted bacterial solutions (106 CFU/mL) at different pH values (pH = 7.4, pH = 5.5) into sterile nitrocellulose Petri dishes (10 mm) placed on agar Petri dishes for 4 days to allow the formation of biofilm on the surface. After applying MN, NPF, and NPF@MN to the biofilm for 24 h, transfer the biofilm to a centrifuge tube containing 5 mL of liquid medium and vortex for 5 min. Colony counting was performed after overnight incubation by diluting the bacteria in PBS and inoculating them on LB agar plates. Additionally, the biofilm was stained using a bacterial live/dead kit.
Pig skins were purchased from reagent dealers to create ex vivo dermal wounds, which were subsequently cut into 5 cm × 5 cm squares with a thickness of about 4–6 mm. A spatula was used to create a scar on the skin that was 2 cm long and 1.5 mm deep to simulate an open wound. Next, immediately added 10 µL of bacterial suspension of 106 CFU/mL to each wound and then covered the inoculated wound with PBS-moistened gauze. The skins were treated with control, MN and NPF@MN groups and placed on solid agar plates and incubated in humidified atmosphere of 5% CO2 at 37°C for 24 h. After 24 h incubation, a 4.5 mm skin biopsy was collected from the center of the ex vivo wound model infection area. The extracted biopsy sample was placed in a 15 mL tube containing 5 mL PBS and ultrasonicated for 1 min. Ultimately, the bacterial solution was serially diluted and inoculated on LB agar plates to quantify the number of viable bacteria remaining in the wound.
5.17. In vitro DU model establishment
400 µM MGO was added to the culture medium of HaCaT cells for 48 h to construct inflammatory models of DUs. Different interposes were applied to observe the function of NPF@MN in regulating inflammatory cytokines.
5.18. Quantitative Real-time PCR (qPCR)
Total mRNA was collected from the cell suspension or skin tissues of wounded mice on day 9 according to the manufacturer’s instructions. The results were analyzed according to the previous literature methodology[49, 59]. The primers for qPCR are shown in Table S1.
5.19. Diabetic wound models and treatments
Eighteen Male C57BL/6J mice were kept in aseptic conditions at 23 ± 2°C and randomly divided into 6 groups: normal ulcer (NU), DU applied with blank microneedles (DU + MN), DU infected with S. aureus and applied with MN (DU + S. aureus + MN), S. aureus-infected DU treated with NPF nanoparticles (DU + S. aureus + NPF), S. aureus-infected DU treated with NPF@MN (DU + S. aureus + NPF@ MN) and the positive group treated with rb-bFGF (DU + S. aureus + rb-bFGF) (n = 3 mice/group).
To establish STZ-induced diabetic models, fifteen C57BL/6J mice were placed on a high-fat diet for 2 weeks. Subsequently, they received intraperitoneal injections of STZ solution (0.2 mL in 0.1 M sodium citrate buffer) twice, with injections administered every other day. Mice that satisfied a blood glucose level > 16.7 mmol/L were considered qualified and used for wound punch (four 6-mm full-thickness excisional wounds). S. aureus was applied onto the wounds for 48 h(after punch to induce infection (107CFU and 20µl/wound), then different treatments (300 µg/mL/wound of NPF@MN; 30 ng/mouse of rb-bFGF) were conducted for 7 consecutive days and the wound skin tissues were collected on the day 9 for histological analysis and inflammatory factor detection. The mice were executed, and their livers, spleens and kidneys were taken for histopathological slide. All the above experimental procedures were permitted by the Ethics Committee of Yueyang Hospital of Integrated Traditional Chinese and Western Medicine, Shanghai University of TCM (YYLAC-2021-107-10, Supporting document 1).
5.20. Histology and immunohistochemical staining
The tissues were collected from mice and were stained with hematoxylin and eosin (H&E) and Masson's trichrome staining by the previous method to determine the remaining wound width and the degree of collagen fibers[49].
The tissue sections were deparaffinized, rehydrated, and subjected to IHC staining using the listed antibodies. The experimental results were analyzed according to previous literature reports[49, 59].
5.21.RNA isolation and library preparation
The extraction of total RNA from animal tissues and the method of library construction using the VAHTS Universal V6 RNA-seq Library Preparation Kit were based on previously published literature[49, 59].
5.22. RNA sequencing and differentially expressed genes analysis
Sequencing on an Illumina Novaseq 6000 platform produced paired-end reads with a length of 150 bp for the libraries. Raw reads for each sample were generated, then processed using fastp and the clean reads for subsequent analyses were obtained. Clean reads were mapped to the reference genome using HISAT2. The FPKM (Fragments Per Kilobase of exon model per Million mapped reads) for each gene was calculated, and HTSeq-count was utilized to obtain the read counts of each gene. R software (v 3.2.0) was employed for PCA analysis to assess the biological duplication of samples. DESeq2 was applied for the differential expression analysis. Differentially expressed genes (DEGs) were selected based on P-value < 0.05 and |Log2foldchange| > 2. Hierarchical cluster analysis of DEGs was performed using R software to demonstrate the expression pattern of genes in different groups and samples.
Utilizing the hypergeometric distribution, we conducted Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) database enrichment analysis for the differentially expressed genes (DEGs) to identify significantly enriched terms. Terms and pathways with an adjusted P-value ≤ 0.05 in both GO and KEGG analyses were considered as significantly enriched.
5.23. GSEA analysis
GSEA (https://www.gsea-msigdb.org/gsea/index.jsp) was utilized to assess the functional enrichment and evaluate the DE mRNAs. Probes were ranked using signal-to-noise ratio, and statistical significance was determined through 1000 gene set permutations [57, 60]. Enrichment analysis was conducted using the R package "clusterProfiler," and the visualization of the insulin signaling pathway was carried out using the R package "enrichplot."
5.24. Statistical analysis
Data were presented as mean ± SD and analyzed using GraphPad Prism 8.0 software. Statistical analysis involved Student’s t-test for comparing two groups and one-way ANOVA or 2-way ANOVA for multiple groups. A significance level of P < 0.05 was considered statistically significant. ***P < 0.001, **P < 0.01, *P < 0.05
{kind=link}