Materials
The following materials were obtained: Trans-cinnamaldehyde (MFY240F0219, Nantong Feiyu Biotechnology Co. Ltd., Nantong, China), β-cyclodextrin (20200503, Shandong Binzhou Zhiyuan Technology Co. Ltd., Shandong, China). Trans-cinnamaldehyde standard substance (710200011, National Institutes for Food and Drug Control, Beijing, China), cinnamic acid standard substance (110786–201604, National Institutes for Food and Drug Control). Carboxymethyl sodium cellulose (150701, Anhui Shanhe Pharmaceutical Excipients Co. Ltd., Anhui, China), Bayer Aspirin enteric tablets (BJ69523, Bayer HealthCare Manufacturing S.r.l., Milan, Italy). Carrageenan (C1013-25G, Sigma-Aldrich Co., St Louis, MO, USA). Acetonitrile (chromatographic grade, TeleChem Corp., Atlanta, GA, USA) Methanol (chromatographic grade, Shandong Yuwang Industrial Co., Ltd., Shandong, China). Ultrapure water (prepared in the laboratory using a pure water machine); acetone (analytical grade, Nanjing Chemical Reagent Co. Ltd., Nanjing, China); Ethanol (analytical grade, Nanjing Chemical Reagent Co., Ltd.).
Molecular docking studies
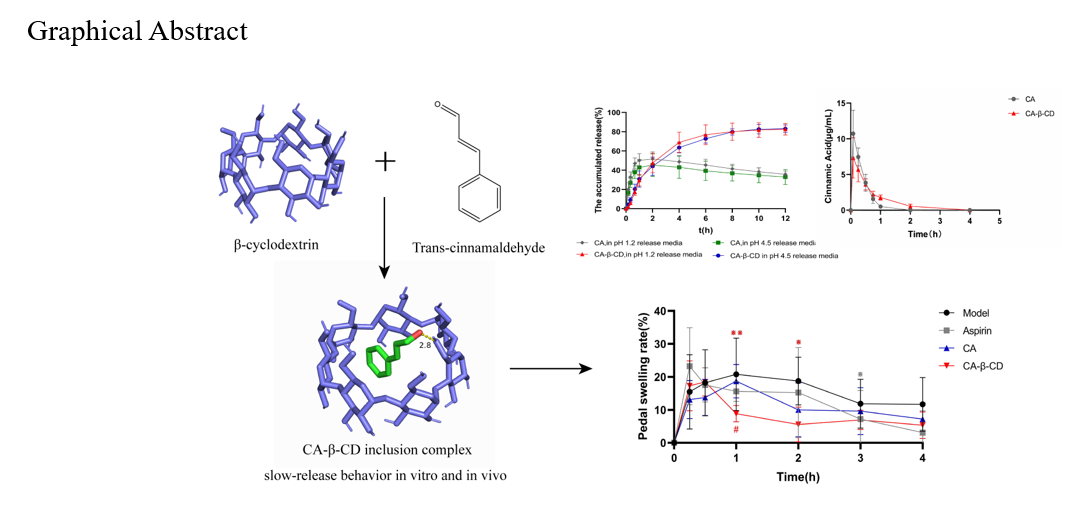
To further elucidate and verify the intermolecular interactions between the β-CD and CA, molecular-level simulations were performed to model molecular docking through theoretical studies to elaborate on the encapsulated binding behavior and obtain reliable structures and properties of the inclusion complexes. The three-dimensional structures of CA and β-CD were obtained from the PubChem database and processed for structure optimization using PyMOL 4.3.0 software. Molecular docking was performed using Autodock 1.5.7 software to simulate and predict the most favorable structures of the guest molecule CA and the main molecule β-CD in the inclusion complex (Fig. 2). The PyMOL software was used to map the conformations of the two bound small molecules [23, 24].
Content determination
Chromatographic conditions: A high performance liquid chromatography system (HPLC) (Waters, Milford, MA, USA) with a photodiode array detector was used to determine the content of CA on an Agilent C18 column (250 mm×4.6 mm, 5 µm) with acetonitrile-water solution (45:55) as the mobile phase, a flow rate of 1.0 mL/min, a detection wavelength of 290 nm, a column temperature of 35℃, and an injection volume of 10 µL.
The sample was prepared by precisely weighing 4 mg of CA-β-CD inclusion, which was then dissolved with methanol in a 10 mL brown volumetric flask, CA was extracted from CA-β-CD by ultrasonic method, and the time was 15 min, finally diluted to 10 mL, fully mixed, and centrifuged at 130000 rpm for 10 min. The resulting supernatant was used to determine the CA concentration using HPLC.
Inclusion complex preparation and physical examination
Preparation
CA and β-CD were accurately weighed according to a molar ratio of 1:1. Cyclodextrin was added to deionized water at a constant temperature of 60°C to prepare the cyclodextrin solution. The CA was dissolved with an appropriate amount of ethanol and the anhydrous ethanol CA solution was slowly injected into the cyclodextrin solution at a constant 60°C with constant stirring by syringe, saturated for 2 h, refrigerated at 4℃ for 12 h, and fully precipitated and filtered. Then the filter residue was washed three times with acetone and dried at 40℃, resulting in a CA inclusion compound (CA-β-CD) [20, 25–27].
Dissolution studies
To evaluate the dissolution profiles of CA and CA-β-CD, a dissolution study (paddle method) was performed. Samples of 100 mg of CA and a weight of CA-β-CD equivalent to 100 mg of CA were placed in a dissolution apparatus. The media used were hydrochloric acid (0.9 L, 0.1 M) at pH 1.2 or potassium dihydrogen phosphate (0.9 L, 0.18 M) at pH 4.5 and 37°C, and they were stirred at a speed of 50 rpm. Samples (2 mL) were removed at predetermined times of 5, 10, 20, 30, 45, 60, 90, and 120 min and immediately supplemented with an equal volume of dissolution medium. Samples were then passed through the membrane, and the CA content was determined using high pressure liquid chromatography (HPLC) (n = 6). The cumulative dissolution rate Mi (%) was calculated using the following equation [28, 29], where Ci is the released concentration of CA at the i-th sample, Vs is the volume before the first sampling (0.9 L), Ci−1 is the concentration of CA at the previous sampling point at time sampling point i (mg/mL), V is the sample volume (2 mL), and W is the total amount of drug delivery (mg).
$${M}_{i}\left(\text{%}\right)=\left[\left({V}_{s}{C}_{i}+V\sum _{n=1}^{i-1}{C}_{i}\right)/W\right]\times 100\text{%}$$
1
In vitro release studies
The shaking-bed method was used in this experiment. Hydrochloric acid (pH 1.2) and potassium dihydrogen phosphate (pH 4.5) solutions were used as the release media, and a dialysis bag with a molecular retention capacity of 1000 D was selected. Samples of CA (10 mg) or CA-β-CD (equivalent to 10 mg of CA) were added to the treated dialysis bag. A small amount of dissolution medium was added, and the ends were tightened using sealing clamps until no leakage was observed. Release medium (200 mL) was measured into a 200-mL wide-mouth flask. The temperature was controlled at 37 ± 0.5℃ and the rotation speed at 140 r/min. Samples (2 mL) were taken at 10, 20, and 40 min, and subsequently at 1, 2, 4, 6, 8, 10, and 12 h. After each sampling time point, the medium was promptly replaced with an equal amount of fresh release medium at the same temperature. The samples were passed through a membrane and analyzed using HPLC to determine the CA concentration. The experiment was performed in triplicate. The cumulative release rate, Qt (%), was calculated for all samples using the equation below. Cumulative release rate-time curves were plotted using GraphPad Prism 9.5 software (La Jolla, CA, USA). The curves were fitted using Origin 2021 software [30–32].
$${Q}_{t}\left(\text{%}\right)=\left[\left({V}_{0}{C}_{t}+V\sum _{n=1}^{t-1}{C}_{t}\right)/W\right]\times 100\text{%}$$
2
C t is the released concentration of CA at time t (mg/mL), V0 is the volume before the first sampling (200 mL), Ct−1 is the concentration of CA at the previous sampling point at time sampling point t (mg/mL), V is the sample volume (2 mL), and W is the total amount of drug delivery (mg).
Stability inspection
To investigate the stability of CA-β-CD at room temperature, six batches of CA-β-CD were prepared simultaneously and stored in airtight glass vials, three of which were stored at room temperature (23 ± 5°C) under normal daylight conditions, and the remaining batches were stored at room temperature in the dark. The batches were sampled at 0, 7, 14, and 30 days for HPLC analysis and the cinnamic aldehyde content was used as the index. The retention time curve was plotted using GraphPad Prism 9.5 to investigate the stability of the inclusion complexes.
Characterization of inclusion complexes
Preparation of samples
CA-β-CD was prepared in the same manner as described previously. A physical mixture of CA and β-CD (PM-CA-β-CD) was prepared by accurately weighing CA and β-CD in a mortar at a molar ratio of 1:1 and grinding them thoroughly to mix.
SEM
Small amounts of β-CD and CA-β-CD powders were scattered and dispersed on small pieces of double-sided tape that were fixed to the surface of a short aluminum rod. The particle shapes and surface characteristics of the samples were measured and photographed using SEM at 20 kV (Apreo 2, Thermo Fisher Scientific, USA).
XRD
Samples of 100 mg each of β-CD, PM-CA-β-CD, and CA-β-CD were analyzed using XRD (Smartlab9, Rigaku, Japan). The samples were mounted in an X-ray holder from the top in a flat quartz bath and scanned at a speed of 2°/min in a scanning range of 5°–90°.
FTIR
Various samples were examined by FTIR spectroscopy (Nicolet-iS10, Thermo Scientific, USA) using the KBr compression method. Approximately 2 mg of each of CA, β-CD, and CA-β-CD were weighed and mixed with approximately 200 mg of dried KBr powder in a mortar and pestle and then an appropriate amount was added into a press die and pressed into transparent flakes at a pressure of 108 Pa. The infrared absorption spectra were scanned in the wave number range of 400–4000 cm− 1, and each sample was scanned three times in parallel.
DSC
Ten mg each of β-CD, PM-CA-β-CD, and CA-β-CD inclusions were taken under N2 airflow, and the samples were accurately weighed in a closed aluminum crimped cuvette (DSC 250, TA Instruments, New Castle, DE, USA) and heated at a rate of 10°C/min in the temperature range of 50 to 300°C.
NMR
A small amount of sample was dissolved in DMSO-d6 for H-NMR and C-NMR detection at 400 MHz power (AVANCE NEO 400, Bruker, Switzerland).
Thermogravimetric analysis
Appropriate samples ranging from 2–10 mg of β-CD, PM-CA-β-CD, and CA-β-CD were placed in an aluminum crucible at a heating rate of 10°C/min. For TG analysis, the reference material used was α-Al2O3 under an N2 atmosphere with a flow rate of 100 mL/min, and a temperature range of 20–800 (NETZSCH STA 449F3, Netzsch, Selb, Germany). To minimize the experimental errors, the test was repeated for each sample [31].
Pharmacokinetic studies
Twelve SD rats (weight, 200 ± 20 g) were randomly divided into two groups, namely, the CA and CA-β-CD groups, and fasted without water before the experiment. After weighing and numbering, the drug was administered via oral gavage at a dose of 50 mg/kg. Approximately 300–400 µL of blood was collected from the orbits at 5, 10, 30, and 45 min and at 1, 2, 4, 6, 8, 10, and 12 h after drug administration. The blood samples were transferred into centrifuge tubes containing sodium heparin, and plasma was isolated by centrifugation at 4000 r/min for 10 min at 4°C. The centrifuged plasma was stored in a freezer at − 80°C for subsequent analysis [33, 34].
The plasma samples were thawed at room temperature (23 ± 5°C), then 100 µL of plasma was precisely aspirated and added to a 1.5 mL centrifuge tube with 35 µL of internal standard salicylic acid solution. Next, 365 µL of methanol was added, and the mixture was vortexed for 30 s, then centrifuged at 13000 r/min for 10 min at 4°C, and the supernatant was extracted. The plasma CA content was then determined using HPLC.
The obtained data were calculated according to the internal standard method to obtain the content of cinnamic acid in rat plasma at different time points for each group. The blood concentration-time curves were plotted by GraphPad Prism 8.0 software and the blood concentration-time data were analyzed by non-atrial fitting using DAS 2.0 analysis software (China Mathematical Pharmacology Professional Committee, Shanghai, China) to calculate each pharmacokinetic parameter. One-way analysis of variance (ANOVA) and least significant difference (LSD) two-by-two comparison tests were performed to calculate the pharmacokinetic parameters based on the concentration-time profiles for each group using SPSS 22.0.
Carrageenan-induced swelling of toes of mice
Twenty-four Kunming mice were randomly divided into four groups: model group (0.5% sodium carboxymethyl cellulose solution), positive drug group (aspirin), CA group, and CA-β-CD group, with six mice in each group. The mice were fasted without water before the experiment. The dose for the positive drug group was 200 mg/kg, while the dose for the CA and CA-β-CD groups was 100 mg/kg CA, administered for 15 min. After administration, 50 µL of 1% carrageenan was injected subcutaneously into the toe of the right hind limb of each mouse to establish an acute inflammation model, and the toe circumference was measured before and 15, 30, 60, 120, 180, and 240 min after injection, and the degree of toe swelling was calculated. All data were expressed as means ± standard deviations (SDs), and statistical analysis was performed using SPSS software (version 22.0; IBM Corp., Armonk, NY, USA). One-way ANOVA and LSD tests were used to analyze and compare data from all experimental groups [35–37].
Toe swelling was calculated as (post-inflammatory toe circumference/pre-inflammatory toe circumference)/pre-inflammatory toe circumference × 100 and was expressed as a percentage.
The inhibition rate of foot plantar swelling was calculated as ([average foot swelling in the model group - average foot swelling in the drug administration group]/average foot swelling in the model group) × 100%.
{kind=link}