Construction and characterization of monolayer model catalysts. A Cu-coordinated porphyrin molecule with clear and typical Cu-N4 structure, [meso-tetra(trimethylsilanylethynyl)porphinato]copper(II) (denoted as 1a, Fig. 1b), was synthesized through literature [36] and utilized as the precursor molecules. The Au(111) single crystal substrate was cleaned via cycles of Ar+ sputtering and annealing up to 500°C. Precursor 1a was deposited by organic molecular beam epitaxy (OMBE) at a sublimation temperature about 170°C onto the Au(111) substrates held at room temperature (Fig. 1a and Supplementary Fig. 1). These 1a molecules form a uniform monolayer self-assembly on Au(111). Each 1a molecule appears as a four-leaf-clover-like structure with four bright lobes, which representing the four -SiMe3 (TMS) groups due to their spatial configuration. The assembled 1a layer is detected with an apparent height of 0.30 ± 0.01 nm (Supplementary Fig. 2), indicating the flat adsorption and monolayer configuration. Notably, the distance between two Cu centers in the 1a monolayer is measured to be 1.50 ± 0.03 nm (Fig. 1e).
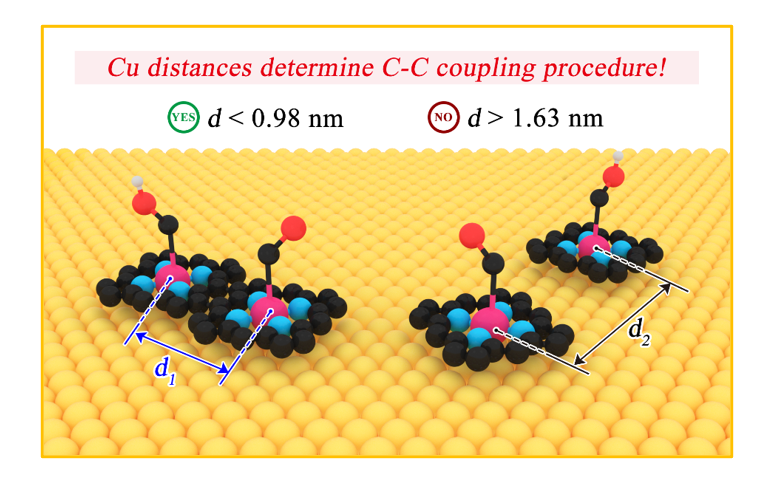
Annealing the self-assembly molecules on Au(111) at 250°C would trigger a chemical reaction of 1a to 1b with a two-dimensional (2D) nanostructure (Fig. 1b). Almost all TMS groups (bright features) were cleaved off and desorbed, in agreement with previous studies [37]. After desorbing TMS groups, the alkyne groups start to form intermolecular five-membered rings with nearby pyrrole ring in porphyrin [38]. These units can further trigger coupling reaction to form chain or square shape conjugated structure (Fig. 1 and Supplementary Figs. 3 and 4). Figure 1g shows the high-resolution image of the 1b layer, which owns a linear fragment composed of a metal center and four porphyrin units. The 1b layer remains the monolayer configuration with a height of 0.18 ± 0.01 nm (Supplementary Fig. 3). The distance between two Cu centers in 1b monolayer is successfully decreased to 0.98 ± 0.03 nm, which is significantly closer than the 1.50 nm of 1a monolayer.
Different from TMS protected alkyne group, we also used another Cu-coordinated porphyrin molecule 5,10,15,20-tetrakis(4-bromophenyl)porphyrin (2a) and Ullmann coupling [39] to generate covalent porphyrin monolayer with longer inter-site distance between Cu centers (Fig. 2a). According to STM images, the vapor deposition of 2a on Au(111) substrate also led to the formation of large area of self-assembled molecules (Fig. 2b). The molecules appear a four-pointed star shape, and each point of the star is corresponding to the Br-phenyl group (Fig. 2c). The height of the assembled 2a molecular layer was measured as 0.28 ± 0.01 nm (Supplementary Fig. 5), and also reflect the flat adsorption and monolayer configuration. The distance between two Cu centers is 1.62 ± 0.03 nm (Fig. 2d). After annealing the self-assembly on Au(111) at 200°C for 30 mins, an Ullmann coupling of 2a via the selective activation of the terminal tran-position Br substituents occurred, bringing extended 2b molecular networks (Fig. 2e-f) [40]. 2b still maintains a monolayer structure after Ullmann coupling (Supplementary Fig. 6), but the distance between two Cu centers increases to 1.73 ± 0.03 nm (Fig. 2g).
Evaluation and comparison of CO 2 RR activity. As displayed above, Cu-coordinated porphyrin monolayers with different inter-distance of Cu sites were successfully constructed on Au(111) substrate, which could be named as Cu098 (polymer 1b on Au(111)), Cu150 (self-assembly of 1a on Au(111)), Cu163 (self-assembly of 2a on Au(111)) and Cu174 (polymer 2b on Au(111)), respectively. The as-synthesized catalysts could be directly utilized as working electrode in electrolysis, due to the good mechanical strength and electrical conductivity of Au(111) substrate. The CO2 electrocatalytic performance of all samples were conducted in a conventional H-type cell with 0.1 M KHCO3 as electrolyte. The CO2RR activity of four samples were estimated firstly via linear sweep voltammetry (LSV). As observed in Supplementary Fig. 7, Cu098, Cu150, Cu163, and Cu174 all show a significantly increasing geometrical current density in a CO2 environment when compared with that in an N2 environment, suggesting the domination of CO2 reduction reaction rather than the parasitic hydrogen evolution reaction (HER). Nevertheless, the influence of Au(111) substrate must excluded since Au itself is also a widely-reported catalyst for CO2RR. Hence, a pristine Au(111) substrate was also used as cathode to test the CO2RR activity. The LSV currents of in CO2 environment of gold is slightly higher than those of the four samples, possibly because organic molecules partially cover the surface of Au(111) [30]. The gas-phase and liquid-phase products were analysed using online gas chromatography (GC) and nuclear magnetic resonance spectroscopy (NMR). Except H2, CO is the only detected product from CO2RR procedure with a maximum faradaic efficiency (FE) of 22.3% (Supplementary Fig. 8). Therefore, Au(111) substrate would not contribute to the multicarbon products in CO2 electrolysis, consistent with the former reports [26, 41].
To better trace the dynamic evolution of gaseous products, differential electrochemical mass spectrometry (DEMS) with a higher temporal resolution [42] was applied to probe the CO2RR procedure on the Cu098, Cu150, Cu163 and Cu174 electrodes with a negatively going potential scan at 1 mV s− 1 in CO2-saturated 0.1 M KHCO3 electrolyte. The faradaic currents on four samples and ionic currents detected by DEMS are displayed in Fig. 3 and Supplementary Fig. 9. As the potential passes − 0.5 VRHE in the negative going scan, a reduction process is observed in faradaic currents of all the four samples. This is accompanied by the evolution of a signal in the ionic current for m/z = 2, indicating the HER procedure and formation of H2. CO2 starts to consume at ca. 0 VRHE on all the catalysts, along with the synchronous increase of faradaic currents (Supplementary Fig. 9). DEMS measurements of Cu098, Cu150, Cu163 and Cu174 all exhibit a steady increase of CO (m/z = 28) yield in terms of ionic currents, indicating the similar activity for CO production. On the contrary, only Cu098 owns a steady increase of C2H4 yield in terms of ionic current (Fig. 3a, m/z = 26 for the fragment of C2H2+). Nothing other than a weak signal of C2H4 could be observed on Cu150, and no ionic currents of C2H4 are detected on Cu163 and Cu174 (Fig. 3b-d).
GC provides the quantitative analysis of reduction products from CO2RR (Supplementary Fig. 10), which is highly coincide with the DEMS results on four samples. The combined product distribution and FEs of these catalysts are directly summarized in Fig. 4a-b. On one hand, four samples exhibit very similar trends of CO FEs within the applied potential range from − 0.4 VRHE to − 1.4 VRHE. Cu098, Cu150, Cu163 and Cu174 obtained the maximum CO FEs of 26.5%, 27.6%, 26.8% and 25.3%, respectively. On the other hand, the Cu098 catalyst exhibited an appreciable 6.1% FE of C2H4 at − 0.8 V RHE (Fig. 4b), obviously superior to Cu150 with a maximum 1.2% FE of C2H4. For Cu163 and Cu174 catalysts, the FEs of C2H4 dropped dramatically to almost negligible levels. These DEMS and GC data directly demonstrate that the distance between Cu sites significantly determines the selectivity of multicarbon products in CO2RR procedure. With a 0.895 nm inter-distance in former report [10] or 0.98 nm in our research, Cu atoms were close enough to each other to enhance the C − C coupling procedure, thus resulting in C2H4 as a main CO2RR product. The C − C coupling pathway was suppressed to a great extent if enlarging the Cu inter-distance to 1.50 nm, and totally blocked when the inter-distance was increased to 1.63 nm and 1.74 nm, leading to the sharp decline of C2H4 production.
It is noteworthy to emphasize the importance of the monolayer distribution of our Cu-coordinated porphyrin molecules. Metalloporphyrin (MP) and metallophthalocyanine (MPc) are both typical model catalysts for M–N4 active sites [27–29]. However, MP or MPc molecules could easily pile up or agglomerate into large particles due to the strong π-π stacking interaction [34]. Once the molecules are stacked together, it is unlikely to precisely control the distance between M–N4 units. Consequently, contrast experiments were also conducted in this research. Monomeric 1a were dispersed in solution and loaded on the Au(111) substrate via a typical drop-casting method. According to scanning electron microscope (SEM) and energy dispersive X-ray spectroscopy (EDX) mapping images (Supplementary Fig. 11), 1a molecules agglomerate to form approximately 5 nm nanoparticles on Au(111) subtrate, although the 1a concentration in dispersed solutions is extremely low. Moreover, the CO2RR activity of this drop-casted 1a catalyst was also evaluated in 0.1 M KHCO3 electrolyte, and GC revealed the generation of C2H4, CH4, CO, and H2 (Supplementary Fig. 12). Specificlly, the C2H4 FEs of drop-casted 1a catalyst fully exceed those of 1a monolayer throughout the applied potential range, and reach a maximum of 9.6% at − 0.8 V RHE. When 1a molecules agglomerate into nanoparticles, Cu sites are close enough to each other to produce C2H4, and it would be impossible to investigate the relationship between inter-distance and FEs of multicarbon products. On the contrary, Cu150 and Cu098 can maintain a monolayer distribution of 1a and 1b after the electrolysis, which is the prerequisite for comparing the impact of inter-site distance on the selectivity of CO2RR products.
Theoretical calculation and mechanism research. To further confirm our proposed mechanism, density functional theory (DFT) calculations were performed to demonstrate the C2H4 selectivity of CO2RR on different inter-distances of Cu sites. Notably, a typical single-atom system always builds the calculated models of M–N/OX-C sites located in graphene with the fitted structure from XAFS data [26]. In our research, Accurate computational models could be constructed based on the 1a, 1b, 2a and 2b molecular structures (Supplementary Fig. 13–16 ). As reported in former research, the overall total reaction processes of C2H4 are formed in multiple reaction steps and 8e– reductions with H2O as the H+ source [23–24]. The formation of *COOH is the initial obstacle to the CO2RR procedure and the potential determining step for CO production. The formation of the intermediate *COOH are all endothermic on four catalysts with close free energy barriers of 0.68 eV, 0.70 eV, 0.73 eV and 0.78 eV for Cu098, Cu150, Cu162, and Cu173, respectively. Subsequently, the *COOH intermediate on the active sites takes another electron and proton to form the *CO intermediate. Four model catalysts possess the identical Cu-N4 structure with similar coordination environment, which could explain the close free energy barriers in the initial steps.
To produce C2H4, two *CO species can couple directly to from *COCO, or a *CO is firstly hydrogenated into *COH and *COH will be coupe with a desorbed CO. In our model catalysts, the two adjacent *CO intermediates adsorbed on the Cu sites are separated by at least 0.98 nm, obviously not suitable for direct 2*CO→*COCO process [10]. Thus, the intermediate *CO should be furtherly hydrogenated into *COH, and the other *CO intermediate needs to be desorbed before the C − C dimerization. Finally, *CHO would be coupled with the desorbed CO by *CHO + CO→*CHOCO, which is the rate-limiting step (RLS) during the whole process [12–13]. Comparing to those of Cu098 (0.89 eV) and Cu150 (1.63 eV), the much larger free energies for C–C coupling step on Cu162 (1.81 eV) and Cu173 (1.92 eV) implies high-energy barriers for the CO2-to-C2H4 process. In addition, the desorbed CO is difficult to continue coupling with *CHO due to the long distance between Cu centers, resulting in higher CO efficiency on Cu162 and Cu173. These calculation results are completely consistent with the experimental results, and directly demonstrate the crucial role of inter-site distance in the pathway of multi-carbon products in CO2 electroreduction.
{kind=link}