The underlying cause of rapid and progressive liver failure leading to sepsis, MOF and high mortality in ACLF is poorly defined (17–18, 25). Hence, there is need for representative experimental model that mimics the course of human ACLF to decipher the pathophysiological journey of the disease and development of new therapeutic strategies. In the current study, we developed a mouse model of progressive ACLF, the CALPS model that resembles the clinical and histological features of human ACLF (17, 18). This model demonstrates that acute liver injury with endotoxemia in cirrhosis leads to progressive hepatocyte necrosis without liver regeneration leading to liver failure and development of secondary organ injury. This model helped to investigate the hypothesis that perpetual presence of hepatotoxin and intestinal endotoxemia lead to hepatocellular necrosis along with inappropriate and widespread activation of the inflammatory cytokine pathway leading to liver failure and subsequent secondary organ failure/sepsis in ACLF (1, 19, 21–22).
Several approaches have been reported in the past by various groups to develop the ACLF model using D-galactosamine/lipopolysaccharide (LPS) (8–12). Though these models showed histological features for the presence of both chronic and acute liver injury, they failed to recapitulate the whole pathological process of this disease due to high short-term mortality and lack the presence of ascites, encephalopathy and secondary organ dysfunction; common features of human ACLF. It is also difficult to understand in these models, whether the high short-term mortality is due to liver failure or endotoxin induced shock.
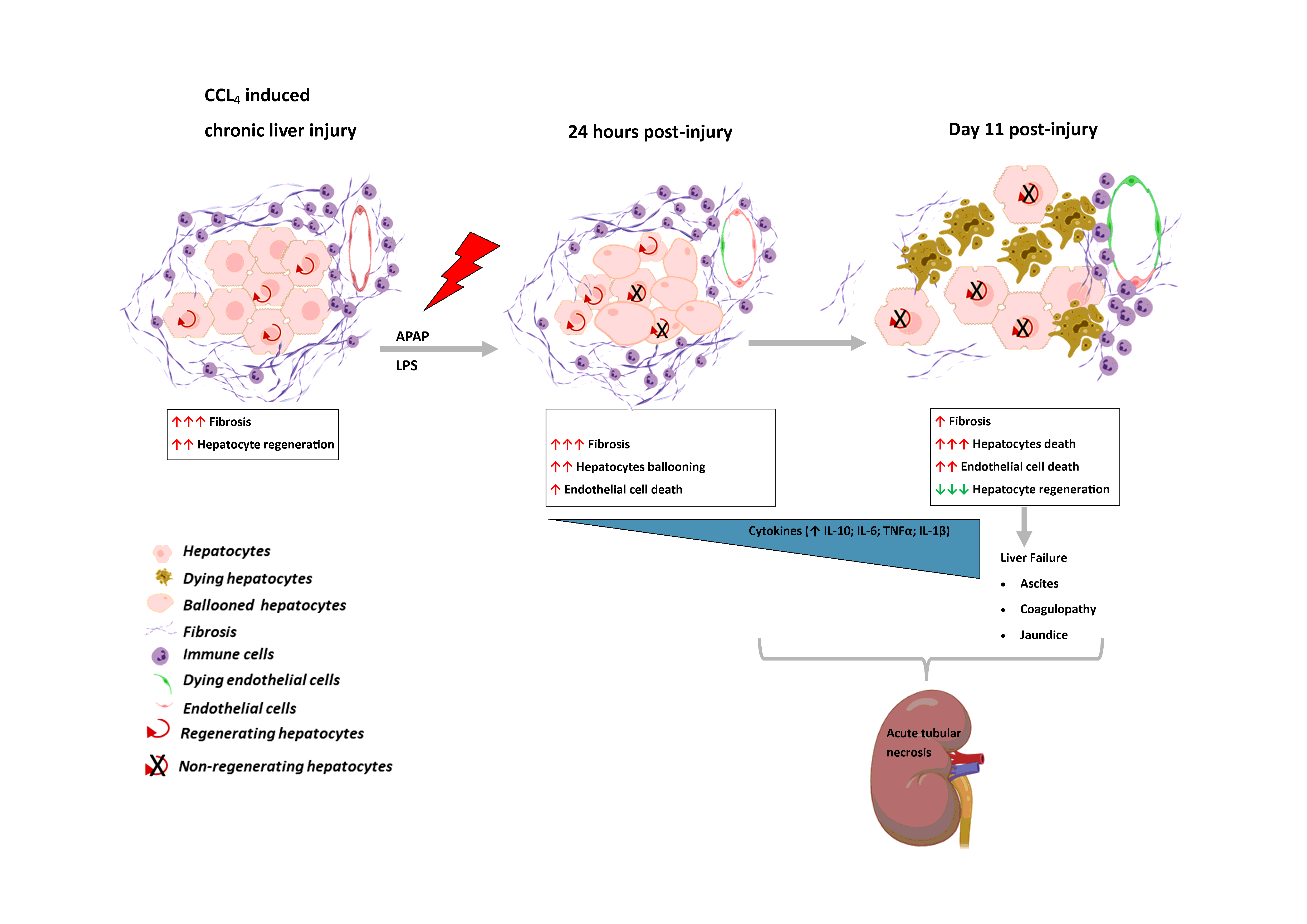
We showed that modest doses of APAP and LPS are sufficient to trigger the liver injury followed by hepatic decompensation (as evident by the presence of jaundice, coagulopathy, ascites and rise in blood ammonia in the current model) and secondary organ injury similar to human ACLF. Our data shows that in response to APAP/LPS injury in cirrhosis liver, there is marked increase in hepatocyte ballooning and endothelial/ immune cell death within 24 hours followed by progressive increase in hepatocyte necrosis from day 7 to 11. This leads to cytokine storm with a persistent increase in pro-inflammatory cytokines (IL6, TNFα and IL-1β) starting from 24 hours and followed by an increase in expression of anti-inflammatory cytokines (IL10) by day 7 post-acute injury with increase neutrophil infiltration. Manifestation of jaundice and coagulopathy followed by the development of ascites and/or encephalopathy post hepatic injury is essential for ACLF as per APASL definition (18). Similar to human ACLF, our animals showed a progressive increase in bilirubin and blood ammonia post APAP/LPS induced liver injury. All animals sacrificed on day 11 showed the presence of ascites with SAAG > 1.1g/dl, hence, conforming to the development of human ACLF. Ballooning degeneration is the precursor of lytic necrosis and has been reported as important histological features in > 50% of ACLF patients (26). The underlying cause of ballooning degeneration is probably a mitochondrial injury followed by impairment of oxidative phosphorylation, ATP-depletion, loss of energy homeostasis and an increase in permeability of the plasma membrane leading to a ballooning of cells (27). It reverses if cells repair the loss of damage or may lead to necrosis if the cells fail to restore the energy homeostasis (27). This raises the possible link between loss of hepatocyte energy and progression of necrosis in response to APAP and LPS in progressive liver injury in ACLF.
Interestingly in the CALPS model, we observed a significant reduction in fibrosis. A reduction in the number of α-SMA + myofibroblasts through apoptosis is a key early event during fibrosis resolution (28). We observed a significant reduction in α‐SMA + myofibroblast post-APAP/LPS treatment. We also observed increased tunnel positive cells in the fibrotic area of the liver; this may be associated with reduction of fibrosis in response to acute injury in the current model. Regression of fibrosis has also been reported in some ACLF patients (18).
The liver has an extraordinary capacity to regenerate on the loss of liver tissue following liver injury due to toxins, surgical resection, infection, or trauma. Compensatory dose dependent increase in liver regeneration with hepatocyte injury has been shown to be associated with spontaneous recovery in various model of drug induced acute live injury (23, 29). This may be quite different in ACLF. In response to APAP and LPS injury, we observed a progressive reduction in hepatocyte proliferation, even though there was a progressive increase in hepatocyte necrosis. Hence, the loss of compensatory regeneration with progressive hepatocyte death indicates impairment in cellular signaling pathways in chronic liver disease which could be responsible for the rapid progression of liver failure in this model of ACLF. Loss of hepatocyte replication has also been shown in the human ACLF (24); however, unlike human ACLF we didn’t observed any increased ductular proliferation, an indicator of HPC mediated regeneration. Indeed earlier we have shown that increased HPCs expansion in ACLF does not contribute to outcome and only hepatocyte replication is associated with spontaneous recovery of these patients (24). Recently, in active infection induced animal model of ACLF, decrease in IL6 and increase in TGF-beta has been shown to be the underlying cause of poor hepatocyte replication. However, we did not find any association of these cytokines with loss of hepatocyte regeneration in our model. In comparison to the cirrhosis, there was a significant increase in hepatic IL-6 within 24 hours post-injury which remained high till day11, while the level of TGF-beta was comparable to that observed in cirrhosis except at 24hrs it decreases (Fig. 2E).
Systemic organ dysfunctions are the common features of ACLF in human (1) of which kidneys are the most affected organs (25). In the present animal model, while there was little discernible change noticed in the kidney/lung histology between day-0 to 7, at day-11, about 80% of the animals showed features of acute tubular necrosis and 30% of them also showed interstitial and/or exudative pneumonia. This suggests that the kidney damage and renal dysfunction are the consequences and not the precipitant of ACLF. It also shows that the renal dysfunction is due to structural damage of kidney, possibly similar human ACLF (30).
In summary, the CALPS murine model of ACLF shows the clinical and histological features of human ACLF in terms of the presence of jaundice, ascites and acute tubular necrosis and renal dysfunction. It showed that perpetual presence of acute liver injury and endotoxemia trigger hepatocyte ballooning followed by massive hepatocyte necrosis in the presence of chronic liver disease. The acute insult in chronic liver disease triggers regression of fibrosis, concomitant impaired hepatocyte regeneration with on-going liver injury results in liver failure and development of secondary organ injury. This model provides the pathophysiological road-map of the development of ACLF in chronic liver disease in response to acute liver damage and endotoxemia. It is also a representative model for understanding the effects of liver failure on renal injury. This model could help in determining the stage specific interventions to prevent development of portal hypertension, renal and lung injury and also to stimulate hepatic regeneration in the presence of on-going liver injury.
{kind=link}