Conventional genetic testing and clinical guidelines focus solely on a small number of well-studied variants or star alleles in pharmacogenes, while the application of NGS techniques provides the possibility to detect a much wider range of (pharmacogenomics) variants. Recent studies have demonstrated that coding variants are rare, population-specific, and with a significant proportion of them potential affecting the protein product (based on in silico assays and metrics) (10–14). At the same time, the role of copy number variants (CNVs) within pharmacogenes (27), as well as variants in non-coding regions, are gaining more attention, with more than 90% of the polymorphisms detected in GWAS pharmacogenomic studies being non-coding (28). Owing to the limited number of thoroughly documented PGx variants and the incredibly large number of identified genetic mutations that should be experimentally validated, the initial evaluation of these variants found must be performed via the use of in silico tools.
The study’s main aim was the assessment of the utility of in silico derived scores, commonly used for variant annotation, toward the characterization of the potential protein function effects of SNVs identified within pharmacogenes. Amongst the assessed algorithms (AdaBoost, XGBoost, Random Forest, multinomial logistic regression), Random Forest presented superior performance and was selected as the final classifier. RFs have been also proven to be robust in the presence of outliers or noise, effective, even without configuration, and useful in cases where the number of available data are limited, compared to the number of available variables, as is often the case with ‘-omics’ data (29, 30).
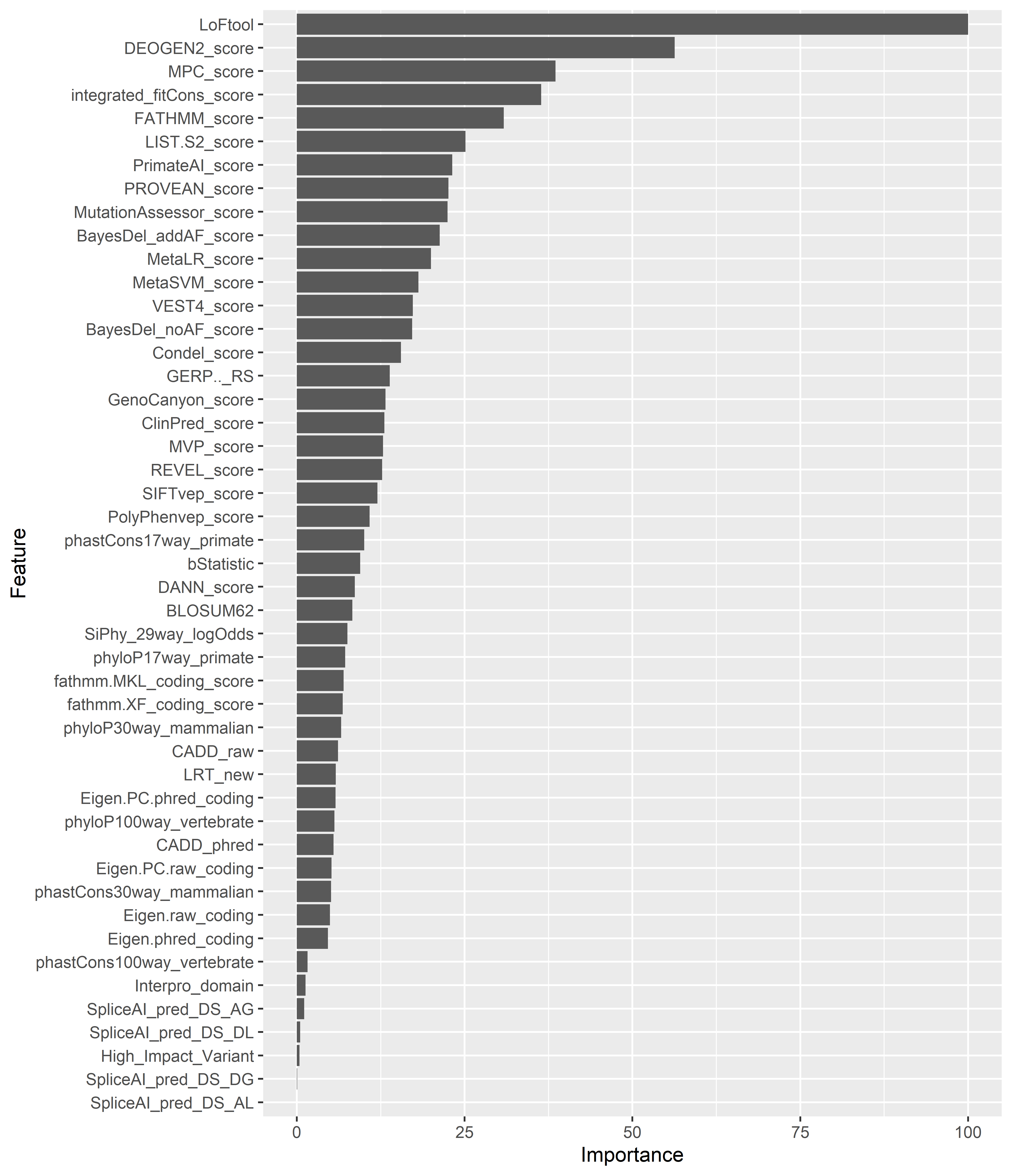
The final classifier required minimum hyperparameter tuning and integrated 7 scores, stand-alone or ensemble ones, and 2 custom created variables. The overall accuracy was found to be equal to 0.85 (95% CI: 0.79, 0.90), with an Area Under the Curve of 0.92 and an Area Under the Precision-Recall Curve (PR AUC) of 0.73. The by-class performance for variants of Normal, Decreased and No Function classes is efficient enough, although there is still room from improvement, especially in terms of sensitivity (0.80, 0.81, and 0.81 respectively). Interestingly, the model appears to be efficient, given the fact that most of the incorporated features are used to distinguish between damaging and benign variants, specifically when it comes to identifying Increased function SNVs. Furthermore, LoFtool, an approach that evaluates the tolerance of a gene to loss-of-function mutations emerged as the most significant determinant of the classification task. Considering the superior performance of the model in identifying “Increased Function” PGx variants, and the observation that this specific class in the training dataset represents only two pharmacogenes, might partially justify the importance of the variable.
Although there is limited published work in this specific area, the possibility of using PGx variants so as to develop classification tools has been previously explored, without however progressing any further due to the limitations and difficulties that accompany this field (31). Firstly, the most frequently examined properties in such classifications tools are the degree of evolutionary conservation, which is observed in lower levels in pharmacogenes (3) and hence its usefulness is debated by a series of studies (26, 31, 32), as well as parameters regarding the structure of the respective proteins, which have been observed to lead to small increases in the efficiency of the classifiers produced (31). Overall, such factors could influence the quality of the output results in classification models, as the one presented herein.
In addition, the training sets used to train computational models are usually comprised of common polymorphisms against variants (mostly SNVs) related to disease-causality, while in terms of drug response, the modifying effect of common polymorphisms cannot be rule out. Moreover, the resulting scores evaluate the pathogenic potential of the examined variants and classify them into two usually categories according to certain applied thresholds. In contrast, PGx researchers usually focus on the induced change in protein function, which can be distinguished at several levels (e.g., increase, decrease, no change, complete loss of activity), while the differential drug response is not a disease, but a phenotype that occurs under specific conditions (i.e., administration of a specific drug).
For example, in a recent study, the adaptation of the proposed classification thresholds and the subsequent integration of selected algorithms, which could provide optimal results for the creation of a comprehensive score, led to a tool with exceptional sensitivity and specificity (26). However, this work focused exclusively on the distinction between loss-of-function and neutral variants, hence ignoring PGx variants that would result in a protein product of increased activity, and which are of interest in PGx field.
The novelty of the present approach lies in the integration of PGx variants characterized as ‘Increased function’; however, the limitations probably affect the performance of the presented classification model More specifically, the collection and curation of larger training datasets with appropriately defined classes for PGx purposes was one of the most significant challenges in this work. Furthermore, the computed metrics demonstrate a difficulty in distinguishing between normal and decreased/no function variants, thus making debatable the suitability of the used features for the characterization of these PGx variants.
Given the challenges and implications for the prediction of functional impact of PGx variants, as well as the complexity of the involved biological processes (33), the findings of this study should be interpreted with caution. For example, discrepancies have been observed not only amongst different algorithms, or between in silico predictions and in vitro activity (34), but also when comparing in vitro and in vivo observations, with the typical example being CYP2D6 * 35, which despite experimental evidence of reduced hydroxylation capacity of tamoxifen(35) has not been associated with reduced activity (36). Moreover, researchers should bear in mind that the same variant may affect the response to different drugs in different ways. For example, although the *10 and *13 CYP2C8 alleles have been found to affect the N-deethylation of amodiacin, the hydroxylation of paclitaxel – which is also metabolized by CYP2C8 - remains unaffected (37).
As mentioned earlier, the presented model has demonstrated promising results, despite the limitations of this computational research field. However, there is still space for further improvements towards a more efficient and robust version of the presented model. More specifically, it would be useful to examine and compare the performance of other machine learning (ML) approaches, supervised or not. Furthermore, significant advantages are expected to emerge from the incorporation of wider training sets, consisting of larger numbers of variants and covering an additional number of pharmacogenes. Emphasis should be also placed on the creation of well-characterized sets of PGx variants at the level of protein effects, both laboratory and clinical, as well as on the improvement of the existing databases to facilitate the export of the requested information. Finally, since the contribution of various factors to the response to a given drug is non-debatable, a more comprehensive approach through systemic genomics would be particularly useful, thus incorporating a variety of different -omics data (38).
{kind=link}