As innovations in prenatal detection, transcatheter and surgical technique, and post-operative management of HLHS have improved survival over the past several decades, focus has expanded to long term neurodevelopmental and quality of life implications in this patient population [6]. In a review of neurodevelopmental outcomes in HLHS from 2007, Wernovsky et al. outlined several studies from the late 1980 to 1990 surgical era that demonstrate intellectual and functional disability affecting over 50% of HLHS patients. Compared to other patients with congenital heart disease including those with single ventricle physiology, those with HLHS have worse neurodevelopmental outcomes [6, 8, 9, 10].
As previously discussed, those with highly restrictive or intact atrial septum continue to have poor survival outcomes despite prenatal diagnosis, planned delivery, and immediate intervention after birth. In a study by Rychik et al. from 2007, 38 patients with HLHS with restrictive or intact atrial septum were identified, of which 16 (42%) had the most severe anatomic obstruction of the atrial septum and 22 (58%) had lesser obstruction. Reversal of pulmonary venous flow with atrial contraction measured by Doppler echocardiography, which has been demonstrated to be a reliable marker for degree of pulmonary venous obstruction with type C pattern representing the most severe form, was utilized to assess degree of atrial level obstruction which correlated with postnatal clinical outcomes [11, 12, 13, 14]. There was no significant difference in percentage of patients with low birth weight, prematurity, or genetic syndromes between the two groups, which is similar to our findings. Early survival was only 38% in the most severe anatomic obstruction group and not affected by prenatal diagnosis (unchanged from observations by the same author from a decade prior). The study noted that early outcome was comparatively better for patients with less anatomic obstruction which likely represents a different group from our study cohort, but there was significant late mortality at the time of subsequent palliative operations. Any degree of atrial restriction in utero likely adversely affects lung development which increases mortality in patients who require single ventricle palliation, supported by the late mortality in our 6-year-old patient in this cohort who died from respiratory complications of a viral illness.
In our study of 6 survivors and 8 non-survivors in 14 patients with HLHS-RAS (Table 1), the mean birthweight was higher in the survivor group (2.8 versus 3.2 kg), though all but 1 patient in both groups were over 38 weeks gestational age. We found more genetic abnormalities in non-survivors (0 patients with genetic abnormalities in the survivor group versus 3 patients in non-survivor group). Our findings were in keeping with previously established data that low birth weight and genetic comorbidities may represent higher risk HLHS [15].
Interestingly, our fetal Doppler assessment at initial fetal echocardiogram was quite varied between survivor and non-survivor groups. We expected that there would be more type C fetal Doppler patterns in the non-survivor group, however we found 3 patients with a type C pattern on initial fetal echocardiogram in the survivor group and 1 with a type C pattern in the non-survivor group. The majority of patients in the non-survivor group had a type B pattern on initial fetal echocardiogram. These findings suggest that that initial fetal Doppler pattern can be variable, may change during pregnancy, and may not necessarily predict outcomes in these patients, though it should be acknowledged that our sample size was small and there may be some subjectivity in terms of echocardiographic interrogation and grading. In our non-survivor group, most patients died in infancy (7 patients) and 1 had later mortality (after Glenn), corroborating aforementioned reports that there is both early and late mortality in this high risk group.
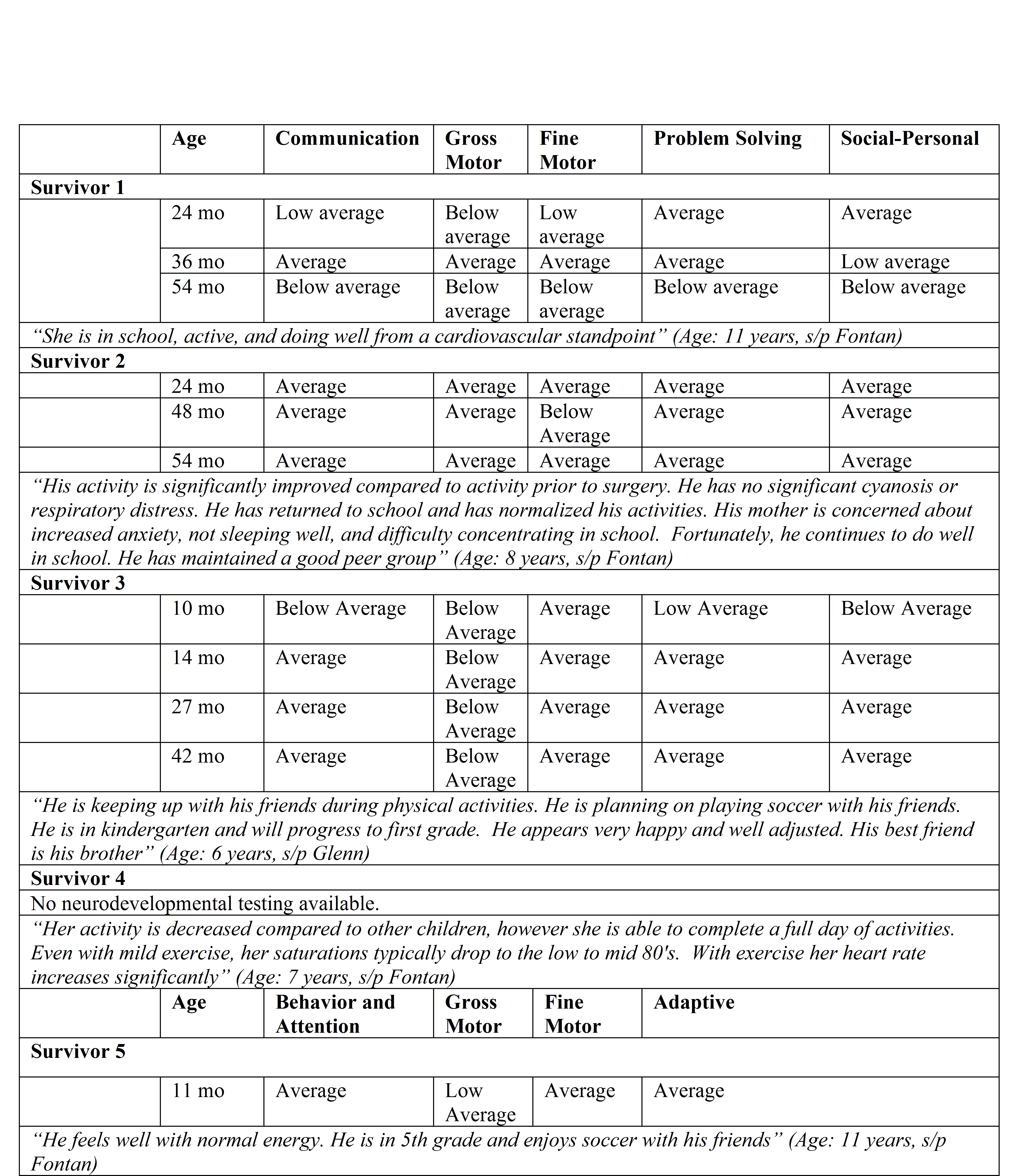
When evaluating long term neurodevelopmental data from our 6 surviving patients at various ages (Table 2), we found that neurodevelopmental milestones were variable at different screening time points. In survivor 1, screening was normal across all domains at 2 and 3 years of age with a plummet across all domains noted around 4–5 years of age. In survivors 2 and 3, neurodevelopmental screening across domains remained relatively consistent throughout early childhood and predominantly in the average to low average range. These findings imply that neurodevelopmental screening varies both individually at different stages of development and between individuals with a common diagnosis, underscoring the importance of universal and serial screening. Moreover, this data shows that those who survive - even within this high risk subtype - may have reasonable neurodevelopmental outcomes in early childhood. Qualitative remarks from the parents of survivors also suggest decent quality of life in survivors in terms of energy, physical activity, and interactions with friends during early school age (6–11 years of age).
Given that HLHS is one of the most common prenatally detected congenital heart disease, being identifiable in the 4-chamber fetal heart view, there is tremendous opportunity for serial prenatal counseling sessions to establish longitudinal care and support for such families [16]. Fetal counseling sessions should include both short term palliative cardiac options as well as long term neurological, developmental, and psychosocial implications to best prepare families for their journey.
Ultimately, our findings are consistent with previous reports of poor survival in HLHS-RAS and forbode the risk for neurodevelopmental delays. However, our data also suggests the possibility of average neurodevelopment even in survivors of the high-risk subtype, and the possibility of reasonable activity tolerance and socialization. Though ambiguity in fetal counseling sessions is cited as a source of stress, it seems reasonable to present the known risks and also depict the range of possible outcomes. Finally, we feel our study contributes to the body of evidence supporting the importance of a robust neurodevelopmental program to maximize the potential of children born with complex congenital heart disease. Future studies with larger sample size are needed to better understand and prognosticate predictors of survival and long-term neurodevelopmental and quality-of-life outcomes within this high-risk HLHS subset.
{kind=link}