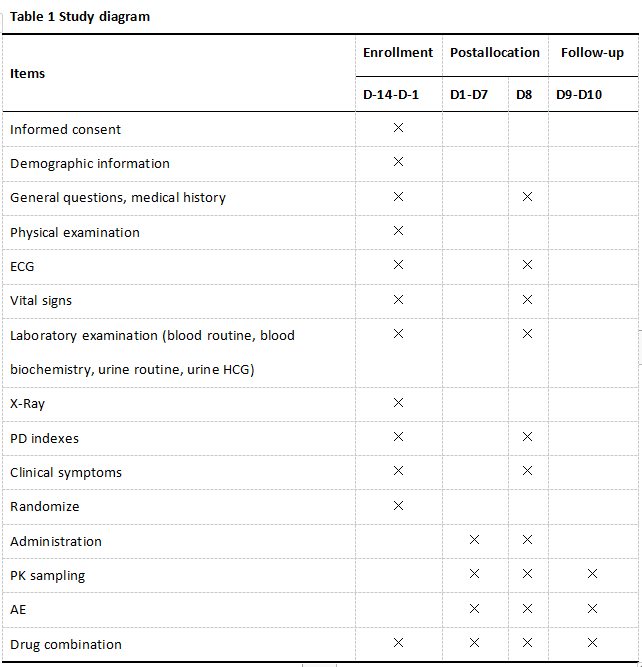
Study design
This is a prospectively planned, blinded, placebo-controlled, parallel-grouped clinical trial with aggregated PPK/PD data. 55 subjects will be randomly allocated into four arms, specifically, 10 of the 55 subjects were selected randomly for the placebo arm who will be orally administered with placebo (Tid), and 45 subjects were randomly assigned to CALAS treatment group (20 mg, 40 mg, 80 mg Tid, 15 subjects per group). Each subject will be randomized to a single dose. The medication, cough and phlegm, body temperature of every subject should be recorded daily during treatment. Each subject will be collected 3-4 blood samples at the following timepointsfor PPK-PPD parameter analysis. Blood samples will be acquired pre-dose (0h) and post-dose administration at 15 min, 40 min, 1 h, 1.5 h, 2 h, 3 h, 4 h, 6 h, 8 h, 12 h, 24 h, 30 h, 48 h after administration. All subjects will receive a laboratory examination and efficacy evaluation on day 8 (Table 1).
Randomization and blinding
According to simple randomization method, 55 subjects are randomized into four groups: placebo: 20 mg : 40 mg: 80 mg group = 10 : 15 : 15 : 15, Furthermore, random coding and drug coding are generated by SAS 9.4 software.
Double blind design is adopted in this trial, that is blind codes that recording drug number and corresponding treatment group are preserved by principal investigator and statistician. Blind codes, folded three times, and sealed in opaque envelopes, which the paste margin is stamped. The blind codes should not be opened during the trial.
Control
Placebo control is adopted in this trial.
Sample size
There are 4 groups in this trial, i.e. placebo group, 20 mg group, 40 mg group and 80 mg group, 55 subjects will be enrolled in the trial, 10 subjects will be randomized in placebo group, 15 subjects will be respectively randomized in 20 mg, 40 mg and 80 mg group.
Study setting
The trial will be conducted in outpatient and Clinical Pharmacology Institution (CPI) at Xiyuan Hospital, China Academy of Chinese Medical Sciences (CACMS). The respiratory physicians will pre-evaluate the patients according to the inclusion and exclusion criteria as follows, and recommend the suitable patients to the study investigators in CPI. All patients voluntarily joined this study with informed consents, and then fundmental indexes including the inquiry, vital signs and physical examination, laboratory examination, ECG and chest radiography will be conducted. The result will be estimated according to the inclusion and exclusion criteria. Eligible subjects will take CALAS on the next morning for 7 days, and participants will return to hospital for safety and effective evaluation on the 8th day. The blood samples will be sent to Shenyang Pharmaceutical University for analysis, and the PPK-PPD data will be analyzed by Beijing Friendship Hospital, Capital Medical University.
Patient and Public Involvement
Patients involvement will be conducted in department of respiratory. The respiratory physicians will pre-evaluate the patients according to the inclusion and exclusion criteria as follows, and recommend the suitable patients to the study investigators in CPI.
Study participants
Inclusion criteria
Participants must fulfill the following inclusion criteria: (1) Aging from 18 to 65 years; (2) In line with the diagnostic criteria of acute trachea-bronchitis; (3) Being less than 48 h of disease course; (4) Being above 5 scores of Bronchitis severity score(BSS); (5) Being informed of the study and voluntarily sign an informed consent.
Exclusion criteria
Subjects who meet any of the following will be excluded: (1) The upper limit of leucocyte exceeds 11*109/L or the neutrophil exceeds 80%; (2) With severe heart disease (acute myocardial infarction or acute myocardial infarction in 6 months), severe cardiopulmonary dysfunction, mental or physical disorders, severe diabetes and immunodeficiency; (3) With severe primary diseases, such as liver and renal hematopoietic system damage, liver function (ALT ≥2 × ULN, AST ≥2 × ULN), kidney function (Cr >1.0 × ULN); (4) With drug allergy history or with allergic constitution, or at least two drug allergies; (5) Pregnant, breast-feeding, and menstruating women, as well as women planning pregnancy within 3 months; (6) Patients who participated in other clinical trials in the last 3 months; (7) Those who cannot take medicine as prescribed by the doctor;(8) People with poor compliance or unsuitability for this clinical trial by investigator's judgement.
Termination criteria
Subject termination
- The investigator may suspend the subject for ethical consideration.
- The investigator may suspend the subject if a serious adverse event (SAE) appears.
- The investigator believes that it is beneficial for subject to withdraw from the trial.
Trial termination
- The principle investigator finds safety problems based on the investigator discussion.
- It is difficult to realize the purpose of the study due to serious deviation.
- The project management unit asks the termination of trial.
Interventions
CALAS is a commercial formulation of A. scholaris leaf with functions of clearing heat, eliminating phlegm, diffusing the lung and relieving cough due to colds and acute bronchitis. Subjects are required to accurately record the medication, cough condition and temperature during the first 7 days, and taking CALAS with 250 ml water under the supervision of the investigators on days 8, The administration time is recorded as "0" hour.
- Dosage regimen:
- 20mg arm: 15 subjects take CALAS (20 mg, Tid) for 7 days.
- 40mg arm: 15 subjects take CALAS (40 mg, Tid) for 7 days.
- 80mg arm: 15 subjects take CALAS (80 mg, Tid) for 7 days.
- Placebo arm: 10 subjects take placebo (Tid) for 7 days.
- Combination therapy
① Antibiotics, western or Chinese medicines with function of relieving cough and reducing sputum are forbidden during the entire trial period, if the patient needs to use the medicine, and the rationality should be evaluated by respiratory physician in advance. All combination should be documented in the case report form (CRF).
② If AE happens, the necessary treatment should be performed according to the subject’s condition, and continuous monitoring should be conducted until the AE restored to a medically acceptable level. All combination and treatment should be documented in CRF.
Sample collection
PK sampling
3 to 4 blood samples of each subject will be collected from the elbow vein and the total sampling need to cover the time-points of 0 h(before last dosing), and 15 min, 40 min, 1 h, 1.5 h, 2 h, 3 h, 4 h, 6 h, 8 h, 12 h, 24 h, 30 h, 48 h after last dosing.
PD sampling
PD samples will be collected at baseline (-1 day) and 8 days after administration.
Sampling collection and storage
2 ml of blood sample is acquired in EDTA anticoagulant tube, before and after administration at different time points, centrifuged at 2000×g for ten minutes at 4℃, and the supernatant is collected and stored under -80℃.
Detection method of PK parameter
The concentration of picrinine, vallesamine , scholaricine and 19-epischolaricinein plasma will be quantitatively analyzed with validated HPLC-MS/MS method.
Detection method of PD parameter
The indexes of SOD, MDA, IgE, Eotaxin, IL-4, IL-10, HMOX1, PDE4B, and PDE4D will be detected by enzyme-linked immune sorbent assay (ELISA) kits, and the ALB will be detected by blood biochemistry.
Proposed trial outcome measures
Primary outcome
(1) Cough disappearance time (clinical efficacy index):
The cough disappearance time of each participant will be recorded to compare the efficacy.
(2) Change in main ingredients of CALAS at different blood concentrations (PK index):
The plasma concentration of four ingredients of picrinine, vallesamine, scholaricine, 19-epischolaricine of the CALAS will be quantitated by HPLC-MS/MS.
Secondary Outcome
- The variations of Bronchitis severity score (BSS), Cough symptom score (CSS), and visual analogue scale (VAS) on day 8, compared with the baseline (Clinical efficacy index).
The BSS, CSS and VAS will be recorded at baseline and the 8th day after administration by respiratory physicians.
- The variations of SOD, MDA、IgE、Eotaxin、IgE、IL-4、IL-10、HMOX1、ALB、PDE4B and PDE4D on day 8, compared with that in baseline (PD index).
The expression of PDE4B and PDE4D, SOD, MDA, IgE, Eotaxin, IL-4, IL-10, HMOX1 enzymes at different time points are detected by ELISA. The content of ALB is determined by serum biochemistry. Moreover the relationship between these indexes and drug concentration will be analyzed.
Safety observation
Routine laboratory tests include hemograms, biochemical indexes (AST, ALT, ALP, GGT, TBIL, DBIL, IBIL, GLU, BUN), urine routine ECG, and urine pregnancy test for women of childbearing age. Analyzing laboratory tests before and after the test for abnormalities, clinical significance, and test drug relevance.
Covariate: Age, BMI, gender, combination, CL, AST, ALT, ALP, BUN, CREA, GGT, TBIL, HB
PD indexes: SOD, MDA, IgE, Eotaxin, IL-4, IL-10, and ALB, HMOX1, PDE4B and PDE4D which were key targets predicted by network pharmacology.
Metabolomics: The blood samples will be detected to explore the characteristic biomarkers embodying endogenous compounds change before and after administration
Adverse events
Each participant will receive safety monitoring throughout the trial. All adverse events (AE) will be reported by investigators, some of them will be collected from spontaneous report of subjects, and some of them will from AE inquiry by investigator on each visit, all the AE details are recorded in AE forms, and the correlation between AE and CALAS will be judged by the investigators. The serious adverse event will be recorded in SAE form, and reported to the Ethics Committee as soon as possible.
Based on previous studies, the expected adverse reactions of CALAS include hiccup, thirst, nausea, excessive sleeping, abdominal distension, increase bilirubin. And the physicians will try their best to prevent and treat AE which brings by CALAS.
Statistical analysis
Primary efficacy index
Cough disappearance time, selecting group as fixed effect and taking baseline value (age, BMI, gender, combination, CL, AST, ALT, ALP, BUN, CREA, GGT, TBIL, HB) as covariates, will be compared between groups by analysis of covariance method.
Secondary efficacy index
The variation of Bronchitis severity score (BSS) , Cough symptom score (CSS), visual analogue scale (VAS) and PD indexes after 7 day dosing, compared with that at baseline are analyzed with ANCOVA method.
Analysis population
The FAS (full analysis set) is defined as the subjects who receive randomization, but not include the subjects whose data is not considered need analyzing by investigator. The SS (safety set) is defined as the subjects who receive at least one medication after randomization. The PKCS (pharmacokinetics concentration set) is defined as the subjects who receive the medication and have at least one time blood concentration data. The PKPS (pharmacokinetics parameter set) is defined as the subjects who receive the medication and have at least one effective PK parameter. For the missing data, no data filling will be performed.
Development of Population Pharmacokinetic/pharmacodynamic model[10-15]
Structure model
One- two- and three- compartmental models are applied to fit the time data of blood concentrations. A proper model will be selected to characterize the drug in vivo behavior according to the objective function value (OFV) and goodness-of-fit plots. The concentration-time data were fitted using the nonlinear mixed-effects modeling software Phoenix NLME (Certara, Inc., Princeton, New Jersey, USA). The first-order conditional estimation method with the η-Ɛ (η: random inter-individual variability; Ɛ: random residual variability) interaction option (FOCE-ELS) was used throughout the model development process.
Inter-individual variation model
The exponential model will be applied to characterize the inter-individual variability (IIV) of population pharmacokinetic parameters:
Pi = P · eηi Equation 1
where Pi and P respectively represent the ith individual value and the typical value for the pharmacokinetic parameter. The relationship between the ith individual and the population values is described using ηi, which is normally distributed with a mean of zero and a variance of ω2.
Intra-individual variation model
The residual error is described by the multiplicative error model:
Ci = C · (1 + εi) Equation 2
where Ci and C accounts for observation and prediction, respectively. εi means the residual error of the predicted value, which is normally distributed with zero mean and a variance of σ2.
Population pharmacokinetic model
The subject’s covariates (Age, BMI, gender, combination, CL, AST, ALT, ALP, BUN, CREA, GGT, TBIL, HB) will be analyzed in a stepwise fashion to identify their potential influences on pharmacokinetic parameters. The categorical covariates (gender and combination) are incorporated using indicator variables. All others (continuous covariates) are centered at the median values and will be included in the model in linear ways. The final PPK is established using the forward inclusion-backward elimination approach. A covariate is considered significant when the addition of this covariate resulted in a decrease in the OFV of >6.635 (P < 0.01), and an elimination of this covariate resulted in an increase in the OFV of >10.828 (P < 0.001).
Population Pharmacokinetic/Pharmacodynamic model
Based on pharmacological action of Alkaloids on the SOD, MDA, IgE, Eotaxin, IL-4, IL-10, and ALB, HMOX1, PDE4B and PDE4D, the relationship between blood drug concentration and PD indexes will fit using an integral PPK/PD model. The indirect response (IDR) model will be used to develop pharmacodynamic model. All the PD indexes are produced with a zero-order input rate and dissipated with a first-order output rate. The rate of change of the response over time with no drug present can be described by
dR/dt = Kin – Kout · R Equation 4
where R is the measured PD index, Kin represents the zero-order constant for production of the response and Kout defines the first-order rate constant for loss of the response. It is assumed that Kin and Kout fully account for production and loss of the response.
According to the action mechanism, drug concentration may stimulate (Equation 7 and 8) or inhibit (Equation 9 and 10) the PD indexes
S(t) = 1 + Emax · C / (EC50 + C) Equation 5
I(t) = 1- C / (C + IC50) Equation 6
where C is the drug concentration. Emax represents the maximum effect attributed to the drug and EC50 represents drug concentration producing 50% of the maximum stimulation achieved at the effect site. IC50 is the drug concentration which produces 50% of maximum inhibition achieved at the effect site
dR/dt = Kin S(t)– Kout · R Equation 7
dR/dt = Kin – Kout · S(t) · R Equation 8
dR/dt = Kin I(t)– Kout · R Equation 9
dR/dt = Kin – Kout · I(t) · R Equation 10
Model verification
Model validation checks the accuracy of the model’s representation of the observation through bootstrap and visual predictive check (VPC). A bootstrap method involved repeated re-sampling with a replacement from the measured data for 1000 times, and the final model will be fitted to the bootstrap datasets. The values of bootstrap parameters [median and the 95% confidence intervals (CIs)] are compared with the parameters of final PPK/PD model. The VPC uses 1000 times Monte Carlo simulation to generate concentration-time profiles. To evaluate the predictive performance of the final PPK/PD model, the observed data will compare with the 5th, 50th, and 95th percentiles of the simulated data.
Interim safety analysis
Interim analysis will not perform in this study.
Data management
The paper medical record and Microsoft excel database are used, data manager creates database according to CRF, single data entry and double check will be conducted by authorized working staff. The data manager draws up the data validation plan, checks the primary and secondary therapeutic index, sends data query form to investigator to explain the problems, until all data queries are cleaned up.
{kind=link}