Effect on storage quality of H. undatus
The H. undatus fruits of both groups were in excellent condition at the beginning of storage (Fig. 1). After 159 h of storage, the squamas of the fruits in the control group were completely dry and brittle, the color was dim and the fruit bodies were significantly corrupted with plaque and were inedible (Fig. 1c). In the trypsin group, the squamas were partly dry; however, the peel was bright and clean, and the flesh was edible (Fig. 1d).
The weight loss rate of each group showed a significant upward trend with increasing storage time (Fig. 1e). The fruit in the control group exhibited a relatively large mean decrease in fresh weight of 1.15% weight loss per day, while the trypsin group showed only a 0.81% (trypsin) weight loss per day (Fig. 1e). There was a significant difference between the trypsin and control groups (p < 0.01).
Impact on the level of cell injury
To further investigate the preservation mechanism of trypsin, the difference of injury on cells between control and trypsin group was evaluated in H. undatus. Figure 1f showed that the membrane lipid peroxidation in the control group sharply increased by fourfold after 7 days of storage. The increase of malondialdehyde (MDA) was fully impeded by trypsin. There was a highly significant difference between the two groups by the end of the storage period (Day 7) (p < 0.01).
Impact on the ROS metabolisms of H. undatus
Excess ROS was the major source of cell injury during storage of fruit. Results showed that the levels of O2−and H2O2 in the fruits of the control group increased with storage (Fig. 1 g and h), exhibiting similar trends as did the MDA content. Trypsin entirely inhibited the accumulation of ROS, especially O2− (Fig. 1g and h).
Transcriptomic analysis
Sequencing and de novo Assembly of Transcriptome
To further reveal the hub genes and key pathway of ROS regulation by trypsin, transcriptomic data was analyzed in H. undatus. The two libraries of the control and trypsin groups produced 50,236,685 and 44,897,144 raw reads, respectively (Table 1). The length of a single read was 150 bp. From control and trypsin group libraries, 48,702,393 and 43,456,887 high quality reads were obtained, respectively. Q20 values were 98.31 and 98.33%, respectively. Q30 values were 94.80 and 94.90%, respectively. Also, 224,395 transcripts were composed by high-quality reads using Trinity software. The average length was 1,086 bp. N50 value was 1,982. The length of transcripts ranged from 201 to 15,462 bp. BUSCO score was 64.8%. Figure S1 showed the length distribution of these transcripts. In fact, 78.63 and 72.92% of transcripts have been mapped, respectively.
Functional Annotation and Analyses
Since there was still no reference genome for H. undatus, a total of 131,559 transcripts and 86,808 unigenes were blasted against six public databases (Swiss-Prot, NR, COG, Pfam, GO and KEGG) (E value < 1e-4). 67,506 (51.31% of all) transcripts and 31,756 (36.58% of all) unigenes were annotated using these databases (Fig. S2, Table S1 and S2). Based on COG and NOG classifications, 3,191 or 2,755 unigenes and 8,288 or 6,677 transcripts were assigned into 24 functional groups, respectively (Fig. S3 and Table S3). The number of transcripts was much higher than that of unigenes. Each unigene was spliced by one or more transcripts. All of these unigenes belonging to three different categories, including biological process (BP), molecular function (MF), or cellular component (CC), have been classified (Fig. S4a).
The main functions were gathered in “binding” and “catalytic activity” on level 2 of molecular function classification (Fig. S4b). In the category of biological processes, they were focused on “organic substance metabolic process” (7,758 unigenes, 15.98%), “primary metabolic process” (5,442 unigenes, 15.12%) and “cellular metabolic process” (5,247 unigenes, 14.54%) (Fig. S4c). In the cellular component, cell part (13.01%) and membrane part (12.35%) were the major parts (Fig. S4d). 7,943 unigenes were assigned to 20 second categories belonging to 6 first categories of KEGG pathways (E-value: 1e-4; Identity: 0; Similarity: 0) (Fig. S5 and Table S4).
Analysis of differentially expressed genes with trypsin treatment
A total of 31,756 unigenes were identified and quantified from 86,808 genes (Table S1). The expression levels of these genes have been concluded by using a volcano plot (Fig. 2a and Table S5). The total number of different unigenes identified was 1,703, including 934 upregulated unigenes (red points) and 769 downregulated unigenes (green points), (p < 0.05, fold change (FC) > 1) in the control and treatment groups (Table S5). On the other hand, a total of 1,117 ROS related genes (orange circle) were determined, including 434 upregulated unigenes (yellow circle) and 465 downregulated unigenes (purple circle). Among the 1,117 ROS related genes, 85 genes (green circle) involved in 1,703 genes (blue circle) were expressed to a significantly different extent (Fig. 2b and Table S6).
GO enrichment analyses
The biological functions of the patterns up- or downregulated by trypsin treatment were analyzed by gene ontology (GO)-based enrichment. The top 10 GO terms in the two expression patterns were shown in Table 2 (FDR <10-6). The upregulated enriched GO terms were “oxidoreductase activity, acting on single donors with incorporation of molecular oxygen (GO: 0016701)” and “oxidoreductase activity (GO:0016491).” On the other hand, besides more oxidation/reduction GO terms, “hydrogen peroxide catabolic process (GO: 0042744),” “peroxidase activity (GO: 0004601),” “oxidoreductase activity, acting on NAD(P)H, oxygen as acceptor (GO: 0050664),” and so on, antioxidant or catabolic GO terms, including “peroxidase activity,” “antioxidant activity” or “catalytic activity” were presented in the downregulated pattern (Table S7).
The major ROS related pathways involved in trypsin regulation can be summarized into a schematic representation (Fig. 3). GO terms are related to one another in a directed acyclic graph (DAG), where more detailed terms are described as children of more general terms. For example, the GO biological process “hydrogen peroxide catabolic process (GO:0042744)” is a child of four terms: “single-organism cellular process (GO:0044763),” “hydrogen peroxide metabolic process (GO:0042743),” “cellular catabolic process (GO:0044248),” and “single-organism catabolic process (GO:0044712).” The GO biological process “oxylipin biosynthetic process (GO:0031408)” is a child of two terms: “oxylipin metabolic process (GO:0031407)” and “fatty acid biosynthetic process (GO:0006633).” These in turn have parent terms, as shown in Figure 3, tracing back to the ultimate ancestor, biological process (GO:0008150), the root of the molecular function ontology. In addition, H2O2 catabolic metabolism was only downregulated (Fig. S6). Cell redox homeostasis was shown to be an upregulated GO process (Fig. S7).
KEGG Pathway analyses
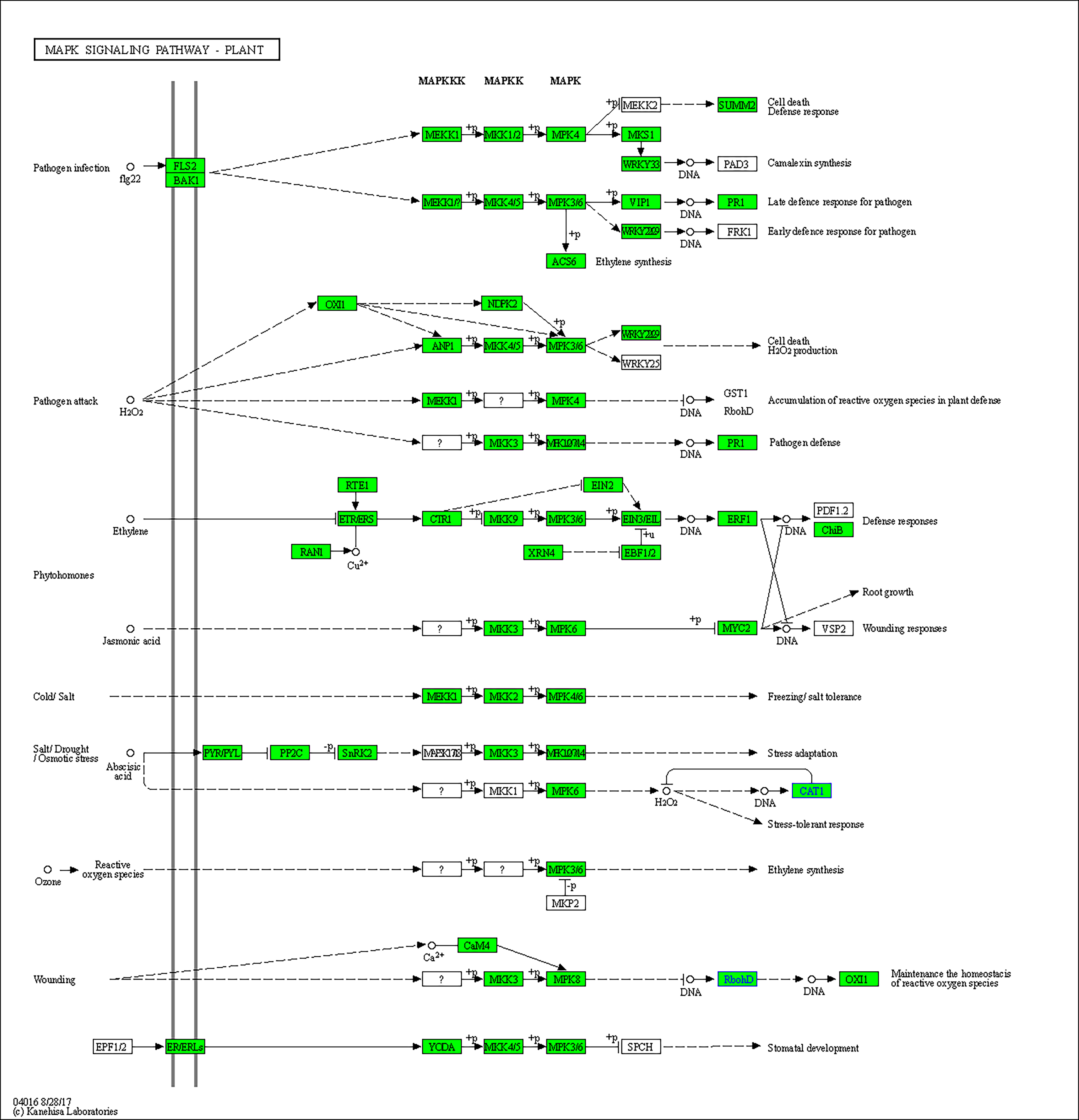
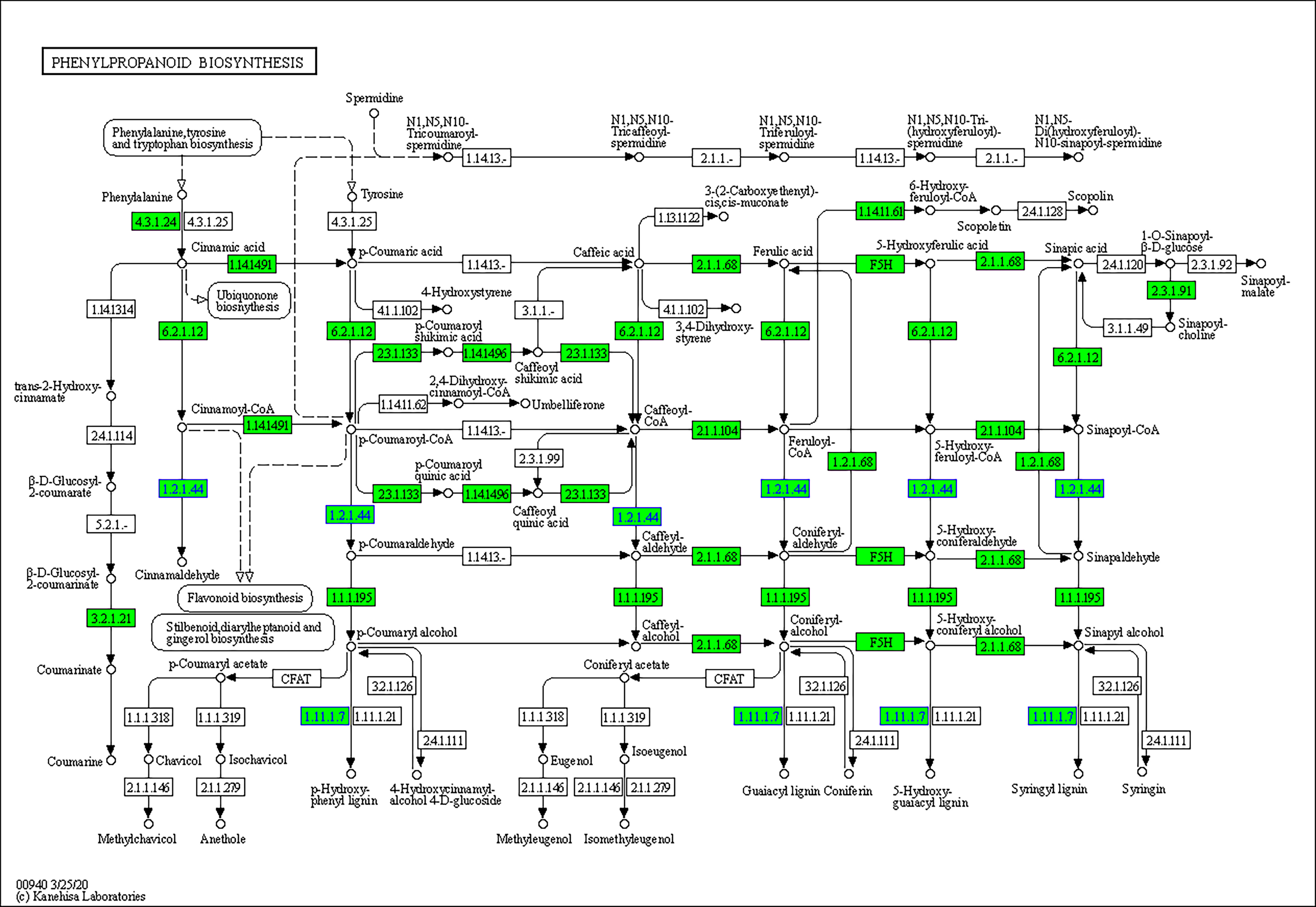
The top 9 enriched pathways were shown in Table 3 (FDR <0.05). With trypsin, downregulated ROS related genes were enriched in several pathways, including the “phenylpropanoid biosynthesis pathway (map00940),” which is associated with fatty acid biosynthesis, as shown in the GO analysis section, “MAPK signaling pathway - plant (map04016),” which involves a series of defense responses induced by ROS, and “Plant-pathogen interaction (map04626),” which highly focus on the hypersensitive response (HR) induced by ROS. On the other hand, “Linoleic acid metabolism (map00591),” “Photosynthesis (map00195),” “Ascorbate and aldarate metabolism (map00053),” and “Porphyrin and chlorophyll metabolism (map00860)” were significantly upregulated.
PPI networks of DERGs
In total, we obtained 85 DERGs (FDR <0.01, 40 upregulated and 45 downregulated) among 1,117 ROS related genes, including 434 upregulated genes and 465 downregulated genes (Table S8).
The PPI subnetwork of total ROS genes was composed of 404 nodes and the first 2,000 edges, containing 40 DERGs (Fig. 4 and Table S9). The upregulated ROS gene PPI subnetwork contained 278 nodes and the first 2,000 edges, including 29 upregulated DERGs (Fig. S8a and Table S10). The downregulated antioxidant gene PPI subnetwork contained 288 nodes and the first 2,000 edges (interactions), including 10 downregulated DERGs (Fig. S8b and Table S11).
Further, the Cytoscape plugin “MCODE” was layered on the PPI network. Twenty clusters were obtained (Table S12). Nodes belonging to the top 6 clusters were labeled by different colors in the PPI network (Fig. 4). The top 6 clusters analyzed by the CytoHubba plugin of Cytoscape were then shown in Figure 5. The central nodes of each cluster were shown in Table 4.
Furthermore, all of the DERGs were selected to construct 3 new PPI networks, including total (60 nodes, 255 edges) (Fig. 6a), upregulated (30 nodes, 81 edges) (Fig. S9a), and downregulated (28 nodes, 62 edges) DERGs subnetworks (Fig. S9b). Three clusters were constructed by MCODE as shown in Figure 6 c, d and e.
Based on the calculation of CytoHubba plugin, because the screen results of either Density of Maximum Neighborhood Component (DMNC) or Maximum Neighborhood Component (MNC) did not match well to the other methods, hub genes were determined by overlapping the genes according to 4 ranked methods, including Maximal Clique Centrality (MCC), Degree, Closeness and Betweenness in cytoHubba (Table S13). Ten hub genes were discovered, including 7 upregulated and 3 downregulated genes (Fig. 6 b and f).
Topological Properties of Networks
The node degree distributions of the total, upregulated, and downregulated ROS related gene subnetworks followed power law fit distributions (𝑅2 = 0.798, 0.765, and 0.762, respectively) (Fig. S10). The subnetwork topological parameters, including network centralization clustering coefficient, and so on, were shown in Table 5.
The subnetworks of DERGs, except the downregulated DERGs subnetwork (𝑅2 = 0.001), were characterized as scale-free networks, even though they were much weaker than ROS related genes networks, which approximately followed power law fit distributions, with 𝑅2 = 0.412 and 0.422, respectively (Fig. S10 and Table 5). Since the DERGs were chosen from the ROS related genes network, either the centralization or the density of the total DERGs network was much higher (0.395 and 0.144, respectively) than that of the total ROS related genes network (0.150 and 0.025, respectively). Finally, the correlation of hub network was decreased to 0.499 (𝑅2 = 0.268).
Accuracy of RNA-Seq Data Verification by RT-qPCR
The accuracy of the RNA-Seq data of ten hub genes of DERGs involved in ROS metabolism were verified by reverse transcription quantitative PCR (RT-qPCR) (Fig. S11). The IDs, NR description, Log2FC(E/C), p value, FDR, and primers of the 10 hub genes are shown in Table 6 and Table S16.
{kind=link}
{kind=link}