Ethics statement
This study was conducted in accordance with the ethical standards, the Declaration of Helsinki, and national and international guidelines, and was approved by the authors’ institutional review board, which adheres to generally accepted international guidelines for animal experimentation.
CircRNA microarray
Five pairs of CRC tumor tissues and corresponding adjacent noncancerous tissues were used for circRNA microarrays. The specimens were obtained from patients undergoing surgery in the Peking University People’s Hospital in 2014; detailed information is shown in Additional file 1: Table S1.
Patients and samples
160 CRC patients who were diagnosed and underwent surgery in Peking University People’s Hospital between 2014 and 2017 were included in this study. Fresh colorectal tumor tissues and matched normal colorectal mucosa tissues were obtained from all the 160 patients. The specimens were obtained and immediately frozen in liquid nitrogen and stored at −80 °C until RNA or protein extraction.
Cellular fluorescence in situ hybridization
The cells were fixed with 4% paraformaldehyde for 15 min and washed with phosphate-buffered saline (PBS) three times for 5 min each time. Further, they were permeabilized with 0.5% Triton X-100 at room temperature for 20 min and washed with PBS three times for 3 min each time. Then, pepsin freshly diluted with 3% citric acid was added and digested at room temperature for 15 min. Subsequently, the nucleic acid fragment was exposed, rinsed with PBS, mixed with 20 μL of a pre-hybrid solution, and pre-hybridized at 50°C for 2–4 h. The hsa_circ_0000231 or miR375-specific probe hybridized at a constant temperature of 50°C. Hybridization was carried out using SSC at 37°C, biotinylated mouse anti-digoxigenin was added dropwise, the fragment was washed with PBS three times for 3 min each time, and the excess solution was absorbed with the absorbent paper. After adding DAPI stain for 10 min, the specimen was subjected to nuclear staining and washed three times with PBS for 3 min each time; the excess solution was absorbed by the absorbent paper. The specimen was sealed with a liquid containing a fluorescent quencher, and the image was observed under a fluorescence microscope.
BaseScope assay
BaseScope assay was performed following the manufacturer's protocols (Advanced Cell Diagnostics, CA, USA). The tissues were sectioned at 5-μm thickness, placed onto Superfrost Plus slides (Fisher Scientific, Loughborough, UK), and allowed to dry overnight at 25°C. The sections were then baked at 60°C for 1 h before deparaffinized in xylene (twice for 5 min) and ethanol (twice for 2 min), and then dried by baking at 60°C for 2 min. Subsequently, hydrogen peroxide was applied for 10 min at 25°C, target retrieval was performed for 15 min at 100°C, and RNAscope Protease III was applied at 40°C for 30 min. The samples were rinsed twice in distilled water between treatments. BaseScope probes (Mm-1700024F13Rik, cat#709881) with positive controls (Hs-PPIB, cat # 701031; DapB, cat # 701011) were then applied, and the samples were incubated for 2 h at 40°C in a HybEZ oven and then with reagents AMP0 (30 min at 40°C), AMP1 (15 min at 40°C), AMP2 (30 min at 40°C), AMP3 (30 min at 40°C), AMP4 (15 min at 40°C), AMP5 (30 min at 25°C), and AMP6 (15 min at 25°C). The slides were rinsed with wash buffer (twice for 2 min) between AMP incubation steps. Finally, they were treated with Fast Red for 10 min at 25°C in the dark, counterstained with Gill's hematoxylin, dried for 15 min at 60°C, and mounted in Catamount permanent mounting medium (Vector Labs, CA, USA).
RNA extraction and quantitative real-time polymerase chain reaction
Total RNA from cell lines and tissue samples was extracted using TRIzol (Invitrogen, USA) following the manufacturer’s instructions. For the plasma, the total RNAs were extracted using an mirVana PARISTM microRNA extraction kit (ABI, USA) following the manufacturer’s protocols. For lncRNA quantification, GAPDH was used as internal control, and PrimeScript RT Master Mix (QIAGEN, Germany) was used for reverse transcription and real-time polymerase chain reaction (PCR). The primer sequences are listed in Additional file 1: Table S2. All reactions were performed in triplicate. The fold change for each gene relative to the control group was calculated using the 2-ΔΔCt method.
Lentiviral short hairpin RNA particles
Recombinant lentiviral particles expressing hsa_circ_0000231 and normal control (NC) were obtained from GenePharm Co., Ltd. (Shanghai, China). SW480 cells were grown to approximately 40% confluence and infected with lentiviral particles in complete medium for 48 h. They were co-treated with the cationic polymer polybrene (8g/ml in water) to increase the infection efficiency. Neither shRNA nor polybrene affected cell viability. Further, shRNA had no off-target effects, and did not affect cell adherence, shape, or viability at the indicated multiplicity of infection.
Cell transfection
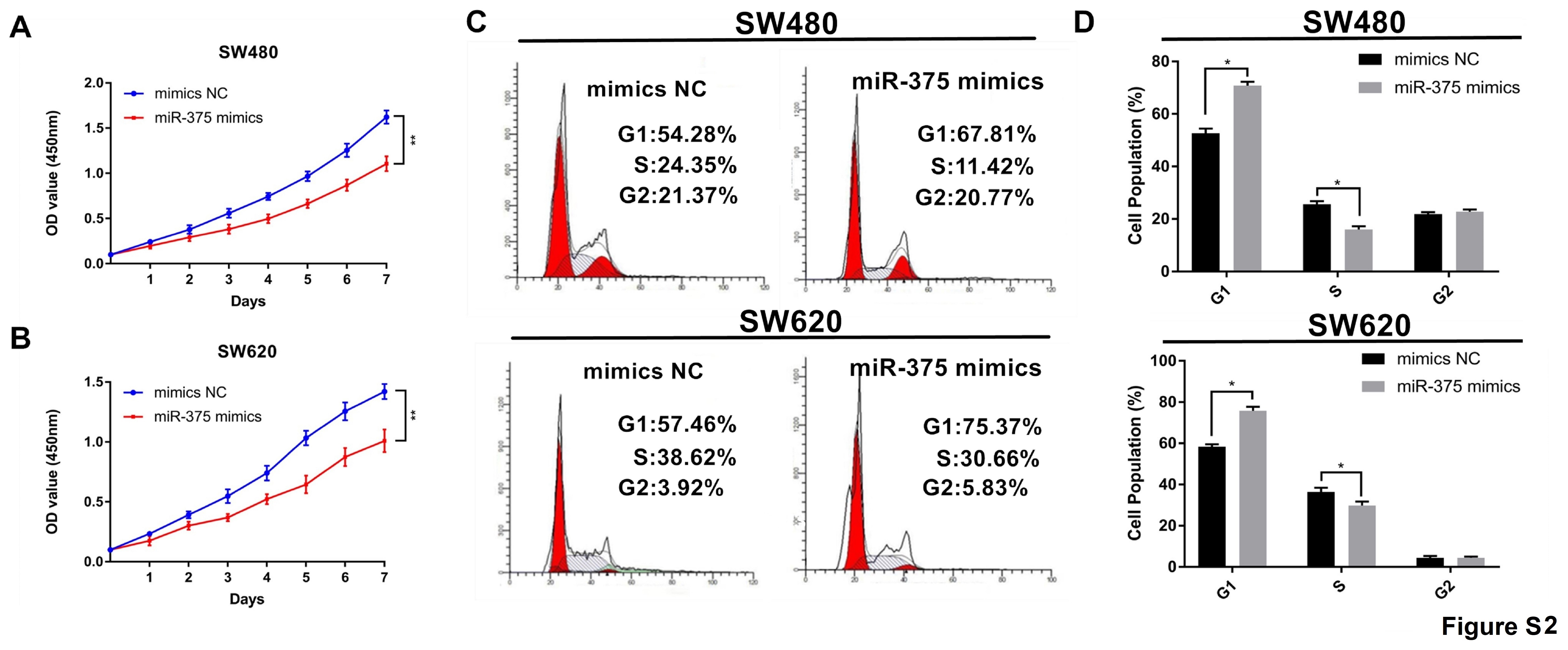
For in vitro studies, siRNA interference sequences targeting hsa_circ_0000231 and the best transcript of ARHGAP12 which hsa_circ_0000231 located at——miRNA (NM_018287) normal control (si-NC) were designed and synthesized (Ribobio, Guangzhou, China) to interfere with the expression of hsa_circ_0000231 or miRNA, which named si-circ_0000231 or si-mRNA, and a final concentration of 50nM was used for transient transfection. Similarly, mimics-NC and miR-375 mimics were designed and synthesized (Ribobio, Guangzhou, China) to overexpress miR-375. Lipofectamine 3000 (Invitrogen, CA, USA) was used for transfection following the manufacturer’s protocols.
For in vivo assays, the hsa_circ_0000231 overexpression cell line was used. The hsa_circ_0000231 gene was cloned into a lentivirus vector LV-GFP-Puro, and SW480 cells were used for infection. Stable transfection cells were established by puromycin antibiotic selection for 7 days, with a concentration of 2.5 µg/ml. The hsa_circ_0000231-overexpressing cells and control cells were named LV-hsa_circ_0000231(LV-circ) and LV-NC, respectively.
Transfection and grouping of cells: Cell transfection were performed in strict accordance with the instructions of Invitrogen's Lipofectamine 3000 Transfection Reagent when the cytoplasm was inoculated with a confluence of about 80%. The Lipofectamine 3000 reagent was diluted with OPTI-MEM culture medium and mixed. The DNA expression plasmid to be transfected was diluted with OPTI-MEM culture medium and mixed with P3000 reagent. The diluted DNA expression plasmid was added in equal volume to each dilution of Lipofectamine 3000 reagent and incubated at room temperature for 5 min. The DNA–liposome mixture was added to the cell suspension and carefully mixed. The culture was continued in a 5% CO2 incubator at 37°C.
Cell proliferation assay
SW480 and SW620 cells (3 × 103 cells) were seeded in complete medium in 96-well plates and infected with hsa_circ_0000231 siRNA. The cell proliferation assay was performed with a Cell Counting Kit 8 (CCK8) following the manufacturer’s protocol, and cell proliferation was detected after 0, 24, 48, 72, and 96 h. The cells in each group were tested for five replicates. The cell proliferation was evaluated by the CCK-8 method using a microplate reader (Molecular Devices, CA, USA) following the manufacturer’s protocols to measure the absorbance.
For the colony formation assay, the transfected cells were seeded into each well of a 6-well plate on day 0 and then incubated for another 14 days. Then, the wells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. The colonies so formed were counted and analyzed using Image J software.
CCK-8 method for detecting cell proliferation curve
The proliferation of CRC cells in each group was detected using the CCK-8 method three repeated times. After transfection for 24 h, the cells were seeded in 96-well plates at 5 × 103 cells/well, and 10μl of CCK-8 solution was added to each well. Then, the cells were incubated in a 5% CO2 incubator at 37°C. After the cells were grown for 2, 3, 4, and 5 days, the absorbance of the cells at 450 nm was measured using an enzyme reader to represent the proliferative activity of the cells during this period. The proliferation curve of each group of cells was plotted, and the difference in the cell proliferation rate was compared between the groups.
Western blot analysis
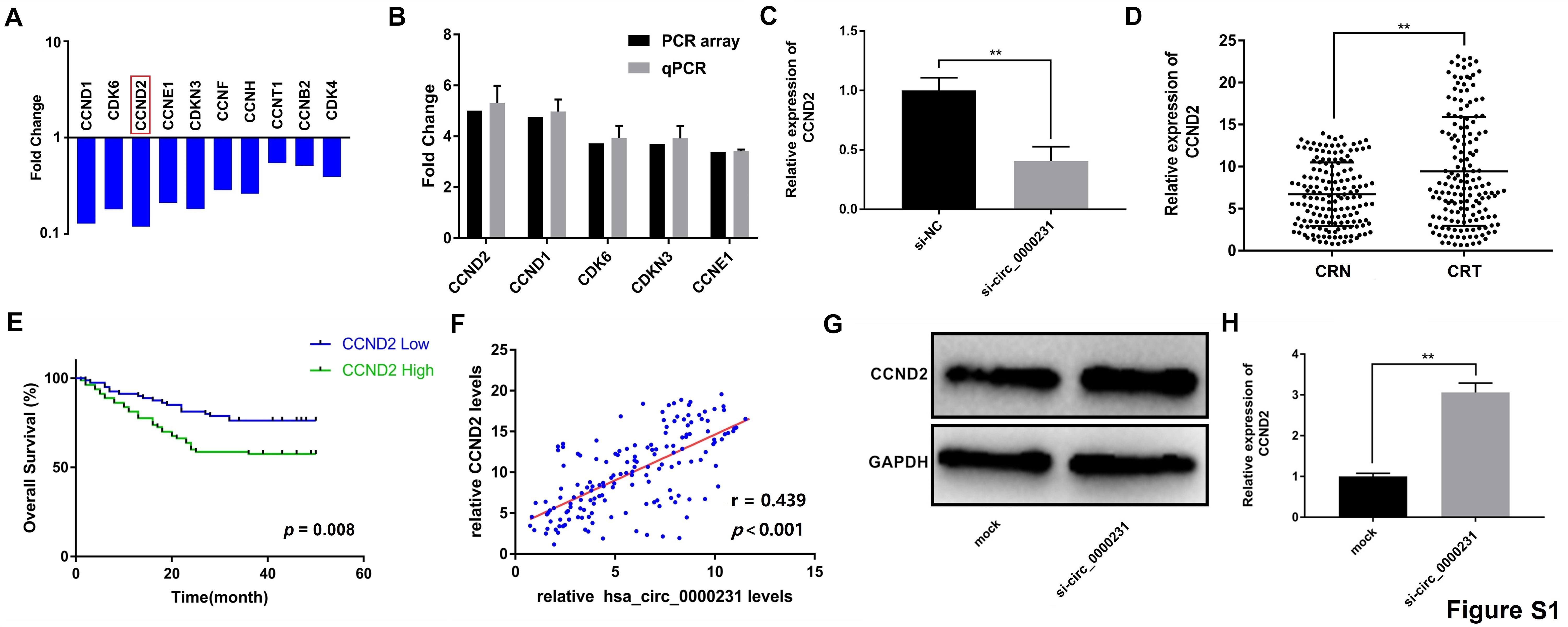
The cells were lysed in RIPA buffer and centrifuged at high speed, followed by protein quantification using a bicinchoninic acid assay three repeated times. The cellular proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were incubated with primary antibodies, followed by a horseradish peroxidase (HRP)-labeled secondary antibody. GAPDH was used as a loading control. The total protein of CRC cells was diluted with RIPA buffer, separated using 10% SDS-PAGE, and then electrotransferred onto a PVDF membrane (Bio-Rad, CA, USA). The membranes were blocked with 5% skimmed milk powder and incubated with primary antibodies against CCND2 (1:1000), IGF2BP3 (1:1000) (Abcam, CA, USA), RB (1:500), and GAPDH (1:5000) (Cell Signaling Technology, MA, USA) at 4°C overnight and then incubated with secondary antibodies (1:5000) (Cell Signaling Technology, MA, USA) at room temperature for 2 h. Finally, the bands were examined by an Immobilob Western Chemiluminescent HRP Substrate (Millipore, MA, USA). The antibodies used in the experiments are shown in Additional file 1: Table S3.
Luciferase reporter assay
The hsa_circ_0000231 and CCND2 fragments containing two putative wild-type or mutated miR-375-binding sites were amplified by PCR and cloned downstream of the luciferase gene in the pGL3 vector (Promega, WI, USA). The constructed reporter vectors were verified by sequencing. Luciferase reporter assays were performed by transiently co-transfecting SW480 cells in 24-well plates with the reporter vectors, miR-375, and the Renilla luciferase construct using Lipofectamine 2000 (Invitrogen, MA, USA). After 48-h transfection, the cells were harvested, and luciferase activity was measured using a dual-luciferase reporter assay system (Promega) and normalized to that of Renilla luciferase.
Biotin-labeled RNA pull-down and mass spectrometry analysis
Biotin-labeled RNA for the linear sequence of has_circ_0000231 and CCND2 was generated by an in vitro transcription reaction with the Biotin RNA Labeling Mix (Roche, Mannheim, Germany) and T7 RNA polymerase (Roche), and then treated with RNase-free DNase I (TaKaRa, Japan). After incubation with the oligonucleotide targeting circular junction, the liner probe was circularized using T4 RNA ligase I and treated with RNase R. After purification with a RNeasy Mini Kit (Qiagen, Inc., CA, USA), the biotin-labeled RNA probe (3 μg) was incubated with cell extracts from CRC cells at room temperature for 2 h and treated with 35 μL of Streptavidin C1 magnetic beads (Invitrogen) for 1 h. After washing, the retrieved protein was detected by Western blot or mass spectrometry analysis (CapitalBio Technology, Beijing, China).
RNA immunoprecipitation
RNA immunoprecipitation (RIP) was conducted with a Magna RIP kit (Millipore, MA, USA) following the manufacturer’s instructions. SW480 cells were harvested 48 h after the transfection of miR-375 mimics or miR-NC and lysed in complete RNA lysis buffer. The cell lysates were incubated with magnetic beads conjugated with anti-AGO2 (Millipore) or negative control immunoglobulin antibody (Millipore) at 4°C for 4 h. The beads were washed with wash buffer. Then, immunoprecipitated RNA and protein were purified and enriched to detect the target RNAs and AGO2 using qRT-PCR and Western blot analysis.
Nude mouse model of ectopic tumors
BALB/c nude (nu/nu) mice, aged 6 weeks old were purchased from Beijing Weitong Lihua Experimental Animal Technology Co., Ltd. Tumors were generated by the subcutaneous injection of 2 × 106 SW480 cells infected with hsa_circ_0000231-overexpressing cells or control lentivirus particles and suspended in 50 L of PBS into the dorsal region near the thigh. Five mice were included in each group. The mice were then weighed and assessed for tumor size every 7 weeks by measuring the tumor length and width. The mice were sacrificed after cervical dislocation, the skin was wiped with 75% ethanol, the abdominal cavity was cut open, and the liver tissues were cut. PMSF (Sigma, MA, USA) (20ug/2ml) were added into the precooled cracking fluid. Take 2ml pre-cooled cracking buffer, quickly add it to homogenizer, and fully grind it under ice bath condition. The samples were centrifuged at 4℃ for 15000rpm for 10min. Take the crude extract of supernatant tissue protein to a new 1.5mL centrifuge tube. The protein was quantified and stored at -80℃.
Statistical analysis
The data generated in this study were all analyzed using SPSS22.0 statistical software. The measurement data were expressed as mean ± standard deviation (x ± s). The two groups were compared using the t test for statistical analysis. The count data were analyzed using the χ2 test. The survival rates were evaluated using the Kaplan–Meier method and tested using the log-rank test. The effects of clinical variables on the overall survival of patients with CRC were determined by univariate and multivariate Cox proportional hazards regression models. Age, T stage, N stage, clinical stage of distant metastasis, and expression of has_circ_0000231 were adjusted for variable analysis in the multivariate Cox proportional hazards regression model. The correlation between groups was analyzed by Pearson correlation, and a P value <0.05 was used as a criterion for statistically significant differences.
{kind=link}
{kind=link}