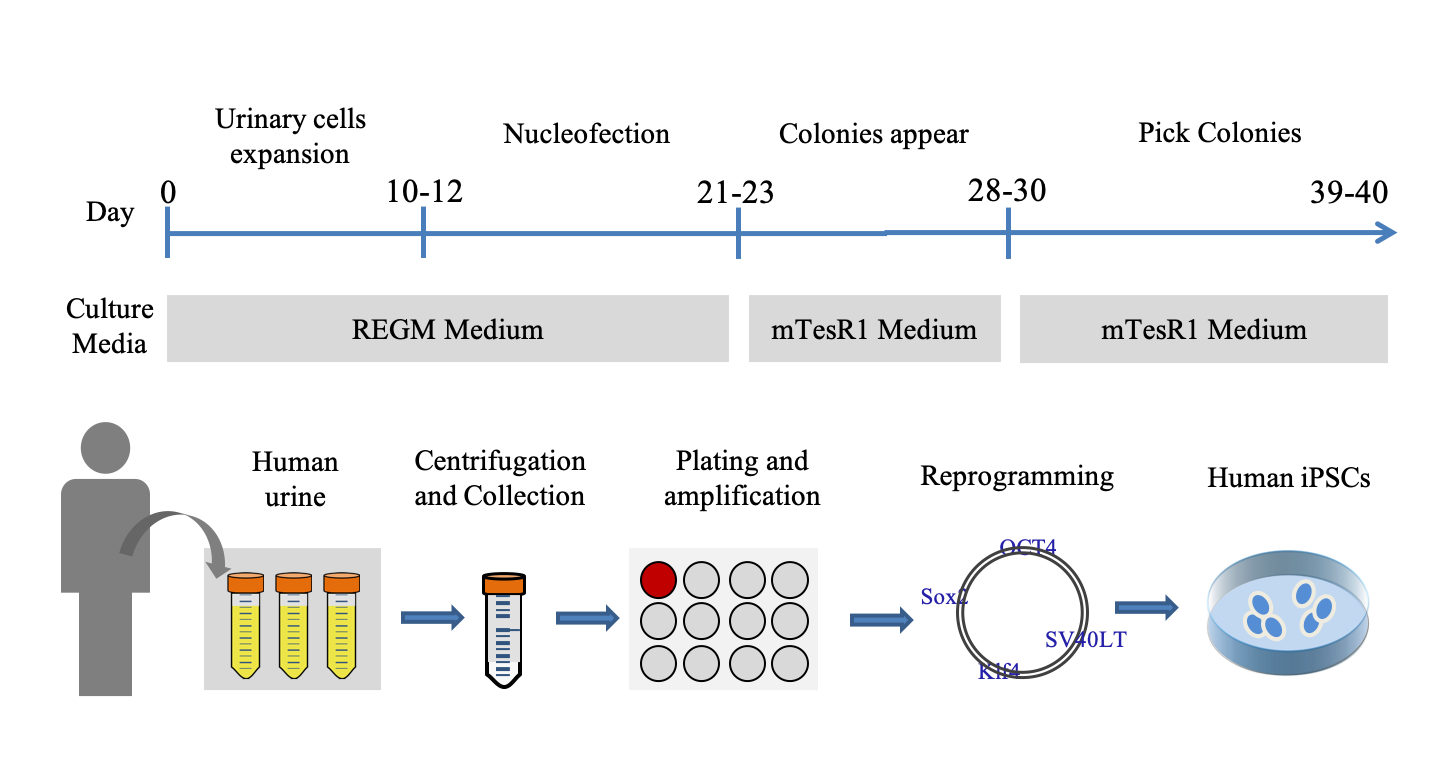
Urinary Cells Collection and Expansion
Zhou et al. detailed the procedure for urinary cells isolation and expansion in 2012[13]. Collect approximate 200ml urine with sterile 50 ml tubes and centrifuge them at 800g for 5 min at room temperature. Gently and quickly pour out the supernatant, so as not to pour out the precipitate. About 5 ml of each tube is left and mixed into a centrifugal tube. Add 20 ml of washing buffer and centrifuge the samples at 800g for 5 min at room temperature. Carefully remove the supernatant, leaving ~0.2 ml plus the pellet. Add 1 ml of primary culture medium to resuspend the cell pellet, and then transfer the volume into a single well of a 12-well plate (coated beforehand with 0.1% gelatin). Add 1 ml of primary culture medium for the first 3 days, but do not remove any medium. Approximately 4 days after plating, remove most of the medium, and add 1 ml of REGM medium (LONZA, CC-3190, Switzerland). Next, change half of the medium every day and observe with a microscope until the cell density reaches 80-90%. Split the cells 1:3 or 1:4 to a 6-well plate and UCs should expand quickly.
hiPSCs derivation and maintenance
The iPSCs were acquired by reprogramming the UCs as described in previous work[13]. The iPSCs were reprogrammed from the UCs of a healthy male of 23-year-old using the same method. For the following experiments, all iPSCs were cultured on Matrigel (BD Biosciences, 356234, USA) in mTeSRTM1 (Stemcell Technologies, 5872, Canada).
Generation of iPSC-derived NSCs through TGF-β/Smad and BMP inhibition
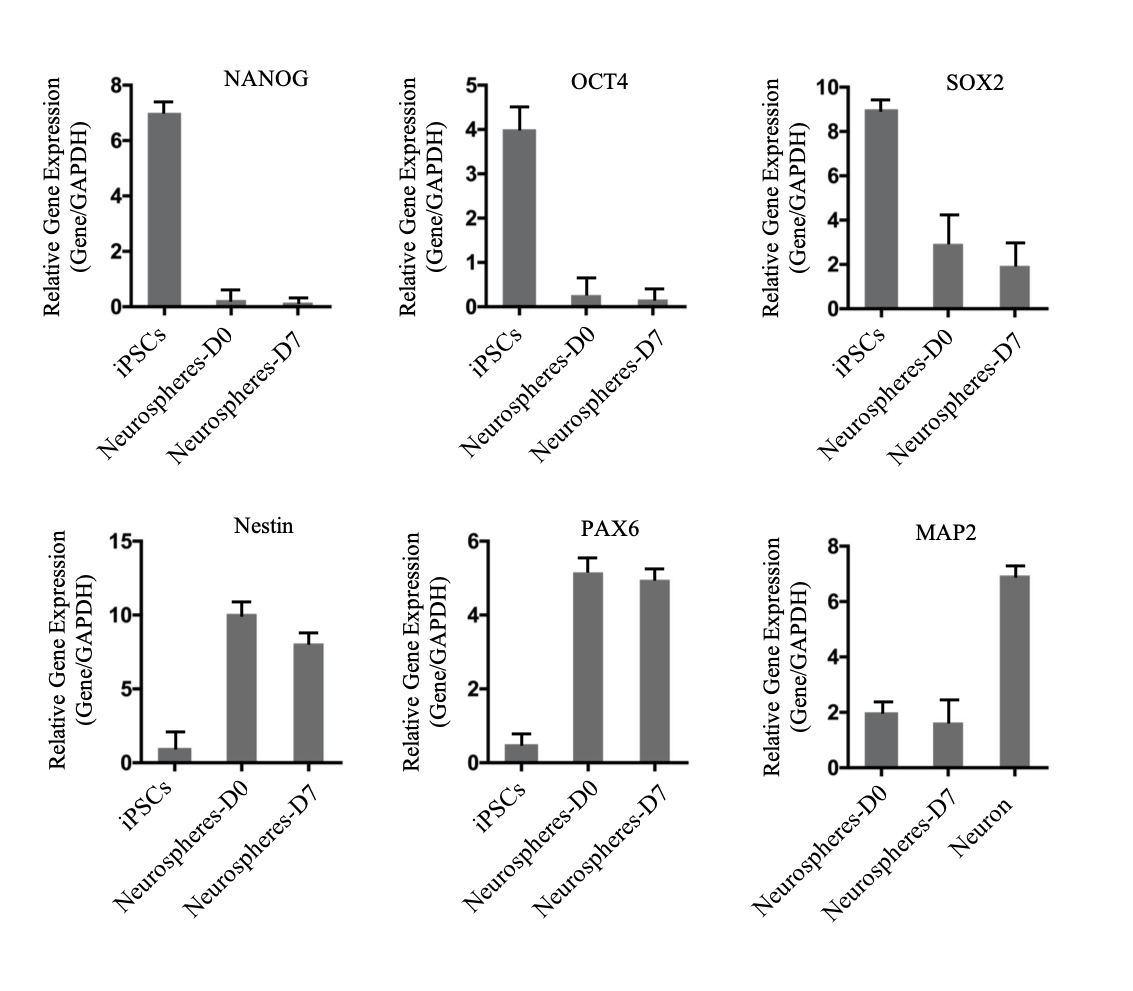
hiPSCs are amplified for 4 days in mTeSRTM 1 medium. Undifferentiated hiPSC colonies were broke into fragments using a P1000 pipette and re-plated onto Matrigel-coated dishes in EBs medium (DMEM/F12, 20% KSR, NEAA (1x), GlutaMax (1x), 0.1% beta-Mecaptoenthano, 5μM SB431542 and 5μM dorsomorphin) to generate EBs for 4 days. The EBs were re-plated onto Matrigel-coated dishes. Neural rosettes were visible and matured within 14 days. Rosettes were picked and then dissociated into single cells with Accutase (Sigma, A6964, USA) that are suspended in culture. After 7-10 days, the single cells produced round neurospheres.
Karyotype analysis
Karyotype analysis was performed in iPSCs at passage 15 and in iNSCs at passage 5. When the cells had reached the logarithmic phase, Colcemid was added to a final concentration of 20 μg/ml for 2 h. The Supernatant was removed, and the pellet resuspended in 8 ml of 0.075 M KCl and incubated for 20 min at 37 °C. The cells were fixed with fresh Carnoy's Fixative (3:1 ratio of methanol: glacial acetic acid). Twenty metaphases were analyzed at 450–500 band resolution using Ikaros (MetaStstems, Germany) on an Olympus BX51 microscope.
Teratoma formation
Human iPSC cells were harvested in 1.5 ml tube and two million cells were injected into the flank subcutaneously and Lower limb intramuscularly of NOD/SCID mice. After 8–10 weeks, euthanasia of mice with rising CO2 levels, tumors were embedded in paraffin, and sections stained with hematoxilin/eosin and histologically analyzed.
Immunofluorescence staining
Cells were briefly fixed in 4% paraformaldehyde for 15 min at room temperature. After permeabilization with 0.5% Triton X-100 in PBS for 5 min, the cells were blocked with 0.5% Triton X-100 and 10% Goat serum for 30 min. Next, the cells were incubated with primary anti-bodiesdiluted in 10% Goat serum at 4 °C overnight and then incubated with secondary antibodies (Supplementary Table.2) diluted in 10% Goat serum for 1 h at room temperature. Nuclei were counterstained with DAPI (Beyotime Biotechnology, C1005, China) for 5 min. Images were acquired with an inverted fluorescence microscope (Olympus, IX73, Japan).
Quantitative Real-time PCR (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen, 15596026, USA). Total cDNA was prepared with HiScript II Q RT SuperMix for RT-qPCR (Vazyme, R223-01, China). qRT-PCR was then performed using specific primers in a CFX96 Real-Time System (Bio-Rad, USA). Primers are listed in Supplementary Table 2.
Electrophysiology.
Electrophysiological recordings were performed at using a whole-cell, voltage- or current-clamp technique. Whole-cell recordings were made with 6 - 9-MΩ borosilicate glass electrodes. Specific protocols were depicted in each figure. An Axopatch 200B amplifier (Axon Instruments, USA) was used to record the electrophysiological signals. The data were acquired and analyzed using Clampfit 10.2 software (Molecular Devices, USA). Borosilicate glass pipettes had resistances of 4–8 MV when filled with a solution containing the following (mM): 140 potassium methanesulfonate, 10 HEPES, 5 NaCl, 1 CaCl2, 0.2 EGTA, 3 ATP-Na2, 0.4 GTP-Na2, pH 7.2 (adjusted with KOH). The bath solution contained the following (mM): 127 NaCl, 3 KCl, 1 MgSO4, 26 NaHCO3, 1.25 NaH2PO4, 10 D-glucose, 2CaCl2, pH7.4 (adjusted with NaOH). Cells plated on coverslips were placed in a submerged recording chamber and were continually perfused with the bath solution equilibrated with 95% O2 and 5% CO2. All electrophysiological experiments were performed at room temperature.
Mycoplasma test
The Lonza MycoAlertTM mycoplasma detection kit was used to estimate the mycoplasma according to the instruction.
Short tandem repeat (STR) analysis
STR analysis was performed on the urine cells and established iPSCs with detection of 21 loci (Amelogenin, D3S1358, vWA, D7S820, CSF1PO, PentaE, D8S1179, D21S11, D16S539, D2S1338, PentaD, D19S433, TH01, D13S317, THOX, D18S51, D6S1043, D1S1656, D5S818, D12S391, FGA) by GUANGZHOU IGE BIOTECHNOLOGY LTD, China.
Data analysis
Statistical analyses for all the experimental data was performed using GraphPad Prism 7 and Microsoft Excel. The data were presented as mean ± SD. Statistical significance were determined using paired T-test.
{kind=link}
{kind=link}