We suggest the possibility to build graphene analogue with the planar hexacoordinate wheel-type Fe@B6H6 cluster as the building block through studying theoretically the geometry, stability and electron structure of its dimer and trimer as well as the dimerization of the two trimers. Employing the dehydrogenation route to polymerization, we can obtain the hexagonal boron sheet that are partly and uniformly filled by Fe atoms in the center of the holes, achieving uniform chemical doping and a very large hexagonal-hole-density. Thus, we may offer a novel cluster-assembled material for experimental chemists to construct graphene analogue.
Research Article
Fe@B6H6 Aggregates: From Simple Building Blocks to Graphene Analogue
https://doi.org/10.21203/rs.3.rs-485747/v1
This work is licensed under a CC BY 4.0 License
You are reading this latest preprint version
Wheel-type clusters
Hexagonal boron sheet
Graphene analogue
Aromaticity
Phonon dispersion
Graphene, the first perfect monatomic two-dimensional carbon crystal, was isolated successfully from graphite in 20041. Its excellent properties, such as extremely high carrier mobility, high thermal conductivity and high specific surface area, have sparked not only the intensive studies on its synthesis and functionalized applications2,3 but also the extensive discoveries towards graphene analogous4,5, consisting of compositions other than carbon. Boron clusters have been found theoretically6–9 and experimentally10–12 to possess a two-dimensional (quasi)planar structure termed boron sheet. However, boron cannot form the stable honeycomb hexagonal-hole framework as graphene because of its electron deficient character. Instead, part of the hexagonal holes need to be filled by boron atoms adopting buckled form so that the boron sheet can maintain its (quasi)planar structure11,12. Therefore, the boron atoms filling the holes serve as an important role in boron sheet and obviously distinguish from the ones forming the hole framework. Under the context, can we choose other atom to partially fill the holes and achieve the better results than that from boron atom? If possible, the more crucial question is how we can fill the holes in boron sheet. The challenging and appealing questions arouse our research interest.
The (quasi)planar hexacoordinate wheel-type clusters, such as CB62– 13 and X@B6H6 (X = V, Cr and Mn)14,15, give us some inspiration to answer the questions. The existence of these clusters indicates that it is possible to fill the hole with atoms other than boron. And then, we can use these clusters as building blocks for constructing graphene analogue through polymerization. As we have shown in our recent work16, the planar wheel-type D6h Fe@B6H6 with good chemical stability is the global minimum isomer and is therefore more attractive candidate for cluster-assembled materials. Here, we studied theoretically the geometry, stability and electron structure of the dimer and trimer of Fe@B6H6 as well as the graphene analogue FeB6. Our calculated results indicate that the structures and charges of the dimer and trimer of Fe@B6H6 are similar to those of the monomer Fe@B6H6. Based on π-electron molecular orbital, Huckel rule and nucleus-independent chemical shifts, the trimer of Fe@B6H6 can be considered as the triphenylene analogue. Moreover, the phonon dispersion indicates that the graphene analogue FeB6 has good dynamical stability. These results indicate that the Fe@B6H6 can be used as the building block to build the graphene analogue FeB6.
The structures of the monomer, dimer and trimer of Fe@B6H6 were optimized by employing B3LYP17,18 method, as implemented in Gaussian 0919, with 6–31 + G(d) basis sets for all atoms except the Fe, which were described by the Stuttgart–Dresden (SDD) effective core potential20. Using the optimized geometries, the HOMO-LUMO energy gap (ΔH−L), vertical ionization potential (VIP)16, vertical electron affinity (VEA)16, natural population analysis (NPA)21 and nucleus-independent chemical shifts (NICS)22 were carried out at B3LYP/6-311 + G(d,p) level. The VIP of monomer, dimer and trimer is defined by the energy difference between the cationic E(monomer/dimer/trimer)+ and neutral E(monomer/dimer/trimer) calculated at the equilibrium neutral geometry, the VEA of monomer, dimer and trimer is the energy difference between the neutral E(monomer/dimer/trimer) and the anionic E(monomer/dimer/trimer)− calculated at the equilibrium neutral geometry, as given in the following equations:
VIP = E(monomer/dimer/trimer)+ – E(monomer/dimer/trimer) (1)
VEA = E(monomer/dimer/trimer) – E(monomer/dimer/trimer)− (2)
For graphene analogue FeB6, its optimized structure is carried out by the DMol3 module implemented in Material Studio 2018 using Perdew-Burke-Ernzerhof (PBE)23 generalized gradient approximation (GGA) and double-numerical properties plus polarization (DNP). In the convergence tolerance, the energy, force, and displacement were set as 10− 5 Ha, 0.002Ha/Å, and 0.005Å, separately24. A vacuum layer of 20Å is added to avoid the influence of periodic adjacent layers. And Monkhorst-Pack k-mesh of 6×6×1 is adopted in Brillouin zone.
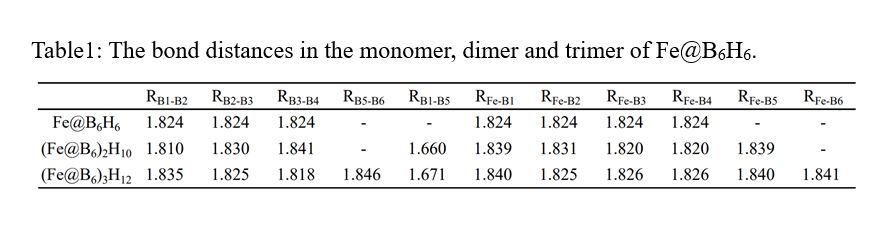
3.1 Optimized structures and some parameters of the monomer, dimer and trimer of Fe@B6H6
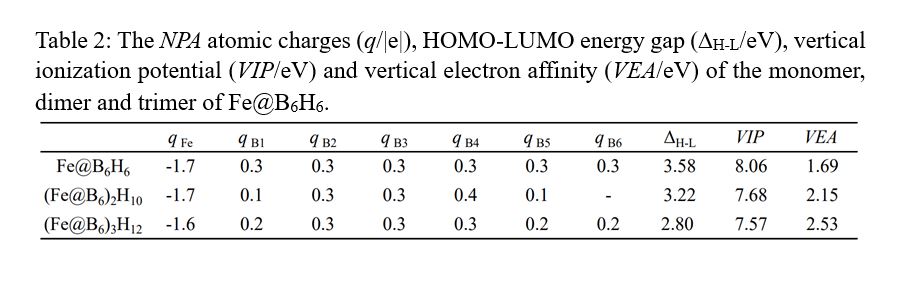
Figure 1 shows the optimized geometries of Fe@B6H6 and the dimer (Fe@B6)2H10 and the trimer (Fe@B6)3H12. The bond lengths and the charges of monomer, dimer and trimer are listed in Table 1 and Table 2, respectively. The monomer Fe@B6H6 forms the perfect regular hexagon with the planar hexacoordinate Fe atom, possessing the highest D6h symmetry. And the bond lengths and charges agree well with those obtained at BP86/6-311 + G(3df,3pd) level16. For the dimer (Fe@B6)2H10,
it has the highest D2d symmetry and the dihedral angle between the two monomer is 90◦ and the distance between them is 1.660 Å which is in good agreement with the experimental value of B-B bond length (1.691 Å)25, suggesting the interaction between the monomers are very strong. While comparing with the monomer Fe@B6H6, the bond lengths and charges the dimer (Fe@B6)2H10 do not change significantly in each monomer except the B atom that links the other monomer. For the dimer (Fe@B6)2H10, all atoms in each monomer are coplanar, which indicates that the character of monomer Fe@B6H6 is well maintained during the dimerization. The dimer (Fe@B6)2H10 with D2h symmetry is a transition state for the conversion to the D2d conformation. For the trimer (Fe@B6)3H12, it has the highest C2v symmetry and the three monomers are perfectly coplanar. The hole in the trimer is not regular hexagon and it is composed of two different types of B–B distances, which are RB1−B5 = 1.671 Å and RB5−B6 = 1.846 Å, respectively.
The ΔH−L, VIP and VEA of the monomer, dimer and trimer of Fe@B6H6 are also listed in Table 2. It can be seen that the ΔH−L values of monomer, dimer and trimer are 3.58, 3.22 and 2.80 eV, respectively. Although the ΔH−L value of trimer is the smallest among them, it is larger than the ΔH−L value (2.63 eV) of triphenylene26, indicating the monomer, dimer and trimer of Fe@B6H6 are chemically stable. The VIP values of monomer, dimer and trimer are 8.06 eV, 7.68 eV and 7.57 eV, respectively, increasing gradually while the VEA values of monomer, dimer and trimer are 1.69 eV, 2.15 eV and 2.53 eV, respectively, decreasing gradually.
3.2 the molecular orbital and aromaticity of the trimer (Fe@B6)3H12
Since Fe@B6H6 exhibits the similar π molecule orbitals to benzene16, the trimer (Fe@B6)3H12 may be the triphenylene analogue. In order to confirm our conjecture, the π-electron molecular orbitals (MOs) of (Fe@B6)3H12 and triphenylene are plotted in Fig. 2. It can be seen that the shape of these Mos of (Fe@B6)3H12 and triphenylene are similar. For example, the HOMO-5 of (Fe@B6)3H12 is bond MO which is similar to the bond MO of HOMO-9 in triphenylene. In addition, both (Fe@B6)3H12 and triphenylene have three degenerate MOs (HOMO, HOMO-6 and HOMO-8 in (Fe@B6)3H12 and HOMO, HOMO-2 and HOMO-6 in triphenylene). As a result, their nine π-electron MOs accommodate 18 π electrons that satisfy the (4n + 2) Huckel rule. Thus, the trimer (Fe@B6)3H12 exhibits the aromaticity and can be considered to be the triphenylene analogue.
NICS is a simple and efficient criterion to characterize aromatic nature. To better understand the aromaticity, the calculated NICS(d) (d = 0 and 1 for inside and above the hole, respectively.) of (Fe@B6)3H12 and triphenylene are also shown in Fig. 2. The NICS(0) = -0.53 ppm and NICS(1) = -0.28 ppm of the hole in the trimer (Fe@B6)3H12 are less
negative than the NICS(0) = -1.72 ppm and NICS(1) = -5.09 ppm of the hole in triphenylene, which indicats that the hole of trimer (Fe@B6)3H12 is less aromatic than that of triphenylene. While the monomer in the trimer (Fe@B6)3H12 has very strong aromatic character since its NICS(1) = -15.2 ppm is more negative than − 9.8 ppm for the monomer in triphenylene, which can compensate the aromaticity of the hole in trimer (Fe@B6)3H12.
3.3 The structure and stability of graphene analogue FeB6
Before building the graphene analogue FeB6, we examined the bigger stable aggregates. Two kinds of different dimerization of the trimer are shown in Fig. 3. The six monomers reveal perfect coplanarity in each of them, indicating the trimer possesses very good ability of plane expansion. Thus, assembling the stable trimers (Fe@B6)3H12 can provide the possibility of building graphene analogue FeB6 as the triphenylene in graphene27,28.
And then, we optimized the graphene analogue FeB6, as shown in Fig. 4. The FeB6 with P6/mmm symmetry is completely planar structure. The boron-ring with Fe atom in the FeB6 has the B-B bond length of 1.860 Å and B-Fe bond length of 1.860 Å, which are slight longer than those (1.824 Å) of Fe@B6H6 monomer. And the bond lengths of two different B-B in the boron-ring without Fe atom are 1.860 Å and 1.661 Å, which are similar to those of boron-ring in Fe@B6H6 trimer. Therefore, the graphene analogue FeB6 preserves the structural features of monomer and trimer of Fe@B6H6.
We also studied the hexagon hole density of the FeB6. The hexagon hole density (η) is defined as26 29a:

According to the formula, the triangular boron sheet has η = 0, the hexagonal boron sheet η = 1/329. For the FeB6, it represents a hexagonal hole density of η = 2/7, which is bigger than those in pure boron α and β29 and very close to the hexagonal boron sheet η = 1/3.
Besides, we also investigated the dynamical stability of the FeB6. The phonon dispersion is shown in Fig. 5. The unit cell of FeB6 monolayer has seven atoms, suggesting that the phonon band structures should have 21 phonon branches. The highest frequency reaches up to 1204 cm− 1, and is higher than the highest frequency of 1036 cm− 1 in BSi30 and 924 cm− 1 in Ti2B231, indicative of robust Fe-B and B-B interactions in FeB6 monolayer. Furthermore, the absence of virtual frequencies at any high-symmetry direction also confirms the dynamic stability of the FeB6.
A proposal that is the possibility of building graphene analogue FeB6 with the Fe@B6H6 aggregates is presented theoretically in the present study. Adopting the dehydrogenation route to polymerization, we can not only place a Fe atom into hexagonal hole instead of B atom in boron sheet, achieving the uniform chemical doping, but also obtain a very large hexagonal-hole-density that is very near to 1/3. The graphene analogue FeB6 await experimental verification because it may possess some novel electronic and chemical properties.
Acknowledgment
This work is financially supported by Scientific and Technological Development Project of Jilin Province, China (Grant No. 20200201104JC).
Funding (information that explains whether and by whom the research was supported)
Scientific and Technological Development Project of Jilin Province, China (Grant No. 20200201104JC)
Conflicts of interest/Competing interests (include appropriate disclosures)
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Availability of data and material (data transparency)
All data generated or analysed during this study are included in this published article
Code availability (software application or custom code)
We own the copyright of Gaussian09 and Dmol3.
Authors' contributions
Chao Wang: performed the data analyses and wrote the manuscript;
Jianhua Hou: contributed to the conception of the study;
Qian Duan: helped perform the analysis with constructive discussions.
- Novoselov KS, Geim AK, Morozov S, Jiang D, Zhang Y, Dubonos S, Grigorieva I, Firsov A (2004) Electric field effect in atomically thin carbon films. science 306:666–669
- Guo S, Dong S (2011) Graphene nanosheet: synthesis, molecular engineering, thin film, hybrids, and energy and analytical applications. Chem 40:2644–2672 S Rev,
- Jiří Tuček (2018) Piotr Błoński.; Juri Ugolotti.; Akshaya Kumar Swain.; Toshiaki Enoki and Radek Zbořil., Emerging chemical strategies for imprinting magnetism in graphene and related 2D materials for spintronic and biomedical applications. Chem 47:3899–3990 S Rev,
- Butler SZ, Hollen SM, Cao L, Cui Y, Gupta JA, Gutierrez HR, Heinz TF, Hong SS, Huang J, Ismach AF (2013) Progress, challenges, and opportunities in two-dimensional materials beyond graphene. ACS Nano 7:2898–2926
- Tan C, Cao X, Wu XJ (2017) Recent advances in ultrathin two-dimensional nanomaterials. [J] Chemical reviews 117:6225–6331
- Boustani I, Quandt A, Hernández E, Rubio A (1999) New boron based nanostructured materials. J chem phys 110:3176–3185
- Evans M, Joannopoulos J, Pantelides S (2005) Electronic and mechanical properties of planar and tubular boron structures. Phys Rev B 72:045434
- Kunstmann J, Quandt A (2006) Broad boron sheets and boron nanotubes: An ab initio study of structural, electronic, and mechanical properties. Phys Rev B 74:035413
- Lau KC, Pandey R (2007) Stability and electronic properties of atomistically-engineered 2D boron sheets. J Phys Chem C 111:2906–2912
- Huang W, Sergeeva AP, Zhai H-J, Averkiev BB, Wang L-S, Boldyrev (2010) A. I., A concentric planar doubly π-aromatic B19 – cluster. Nat chem 2:202–206
- Li W-L, Chen Q, Tian W-J, Bai H, Zhao Y-F, Hu H-S, Li J, Zhai H-J, Li S-D, Wang L-S (2014) The B35 Cluster with a Double-Hexagonal Vacancy: A New and More Flexible Structural Motif for Borophene. J Am Chem Soc 136:12257–12260
- Piazza ZA, Hu H-S, Li W-L, Zhao Y-F, Li J, Wang L-S, Planar hexagonal B36 as a potential basis for extended single-atom layer boron sheets. Nat. commun. 2014, 5, 3113
- Exner K, von Ragué Schleyer P (2000) Planar hexacoordinate carbon: a viable possibility. Science 290:1937–1940
- Li L, Xu C, Jin B, Cheng L (2013) Benzene analogues of (quasi-) planar M@ BnHn compounds (M = V–, Cr, Mn+): A theoretical investigation. J chem phys 139:174310
- Hou J, Duan Q, Qin J, Shen X, Liang Q, Jiang D, Gao S, Planar wheel-type M©B n H n 2–/–/0 clusters (M = Cr, Mn and Fe for dianion, anion and neutral, respectively; n = 6 and 7). RSC Adv. 5, 49, 38873–38879
- Hou J, Duan Q, Qin J, Shen X, Liang Q, Jiang D, Gao S (2015) Unconventional charges distribution in the planar wheel-type M@ B6H6-/0/+(M = Mn, Fe and Co): central M with negative charges and peripheral boron ring with positive charges. Phys Chem Chem Phys 17:9644–9650
- Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J chem phys 98:5648–5652
- Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785
- Frisch M, Trucks G, Schlegel HB, Scuseria G, Robb M, Cheeseman J, Scalmani G, Barone V, Mennucci B, Petersson G (2009) Gaussian 09, revision A. 02; Gaussian, Inc. Wallingford CT 19:227–238
- Andrae D, Haeussermann U, Dolg M, Stoll H, Preuss H (1990) Energy-adjustedab initio pseudopotentials for the second and third row transition elements. Acta biotheor 77:123–141
- Wiberg KB (1968) Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 24:1083–1096
- Schleyer PvR, Maerker C, Dransfeld A, Jiao H, Hommes NJ (1996) v. E., Nucleus-independent chemical shifts: a simple and efficient aromaticity probe. J Am Chem Soc 118:6317–6318
- Ernzerhof M, Gustavo E, Scuseria (1999) Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J chem phys 110:5029–5036
- Li H, Duan Q, Jiang D, Hou J, Guo X (2019) A First-Principles Study of Boron‐Doped Bc2n Sheet as Potential Anode Material for Li/Na‐Ion Batteries. ChemElectroChem 6:3797–3805
- Allen FH, Kennard O, Watson DG, Brammer L, Orpen AG, Taylor R, Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc., Perkin Trans. 2. 1987, S1-S19
- Aihara J-i (1999) Reduced Homo – Lumo Gap as an Index of Kinetic Stability for Polycyclic Aromatic Hydrocarbons. J Phys Chem A 103:7487–7495
- Das S, Irin F, Ahmed HT, Cortinas AB, Wajid AS, Parviz D, Jankowski AF, Kato M, Green MJ (2012) Non-covalent functionalization of pristine few-layer graphene using triphenylene derivatives for conductive poly (vinyl alcohol) composites. Polymer 53:2485–2494
- Chen K, Wan X, Liu D, Kang Z, Xie W, Chen J, Miao Q, Xu J (2013) Quantitative determination of scattering mechanism in large-area graphene on conventional and SAM-functionalized substrates at room temperature. Nanoscale 5:5784–5793
- Tang H, Ismail-Beigi S (2007) Novel precursors for boron nanotubes: the competition of two-center and three-center bonding in boron sheets. Phys Rev lett 99:115501
- J, Dai.; Y, Zhao.; X Wu (2013) J, Yang.; X, Zeng., Exploration of Structures of Two-Dimensional Boron–Silicon Compounds with sp2 Silicon. J Phys Chem Lett 4:561–567
- T B, P L, Y JXu;JZhang, Chen.; O E, F, Wang B (2018) Wang., Hexagonal Ti2B2 Monolayer: A Promising Anode Material Offering High Rate Capability for Li-Ion and Na-Ion Batteries. Phys Chem Chem Phys 20:22168–22178
Due to technical limitations, table 1,2 is only available as a download in the Supplemental Files section.
{kind=link}
{kind=link}