Cell culture and induction of cisplatin-resistant NSCLC (CR-NSCLC) cells
The parental CS-NSCLC cell lines, including A549 (ATCC® CCL-185™), H1299 (ATCC® CRL-5803™) and Calu-3 (ATCC® HTB-55™), were purchased from American Type Culture Collection (ATCC, USA) in Jan. 2019, and cultured in the incubator with standard culture conditions (37 ℃ and 5 % CO2 humidified atmosphere). The cells were authenticated by STR profiling and were identified as mycoplasma-free by a commercial third-party company (Abace Biotechnology, Beijing, China). The Roswell Park Memorial Institute 1640 medium (RPMI-1640, HyClone, USA) containing 10% fetal bovine serum (FBS, Gibco, USA) was used for cell cultivation. According to the experimental procedures provided by the previous work [36, 37] and our preliminary experiments (data not shown), the CS-NSCLC cells were exposed to continuous low-dose cisplatin treatment, ranged from 0.5 μg/ml to 5 μg/ml, for 80 days in a step-wise manner to generate descendent CR-NSCLC cells (A549/DDP, H1299/DDP and Calu-3/DDP). After that, the CR-NSCLC cells were stimulated with high-dose cisplatin (25 μg/ml) for 0 h, 24 h, 48 h and 72 h, to validate the successful induction of CR-NSCLC cells.
Vectors transfection
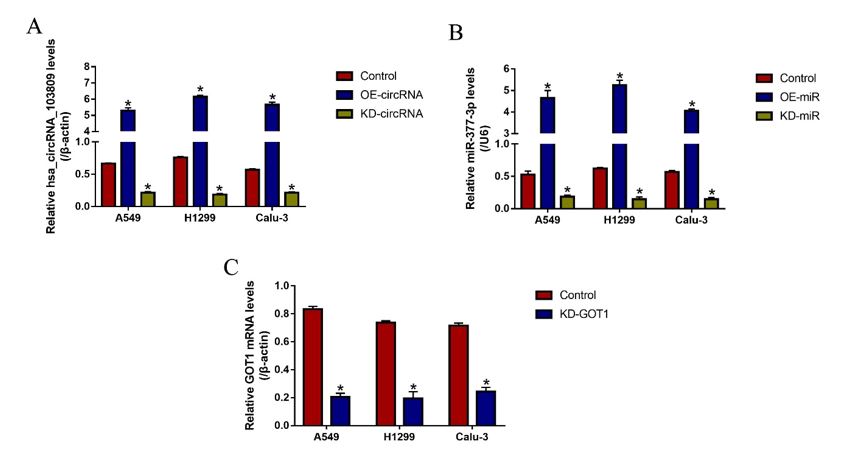
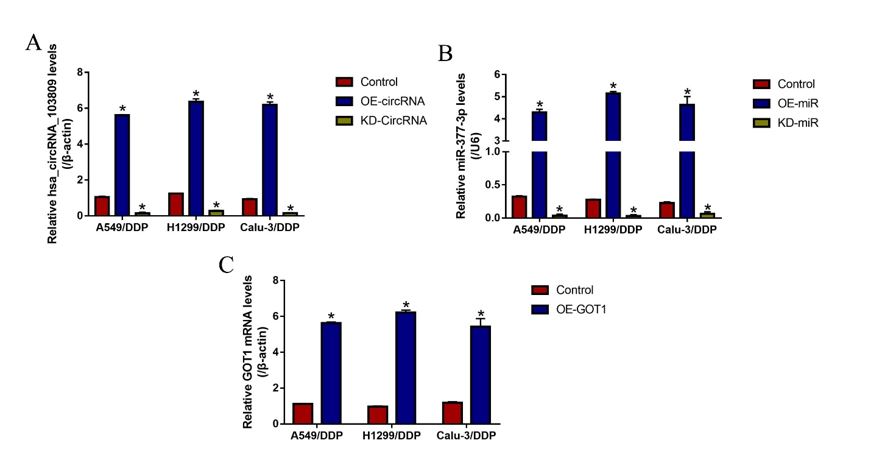
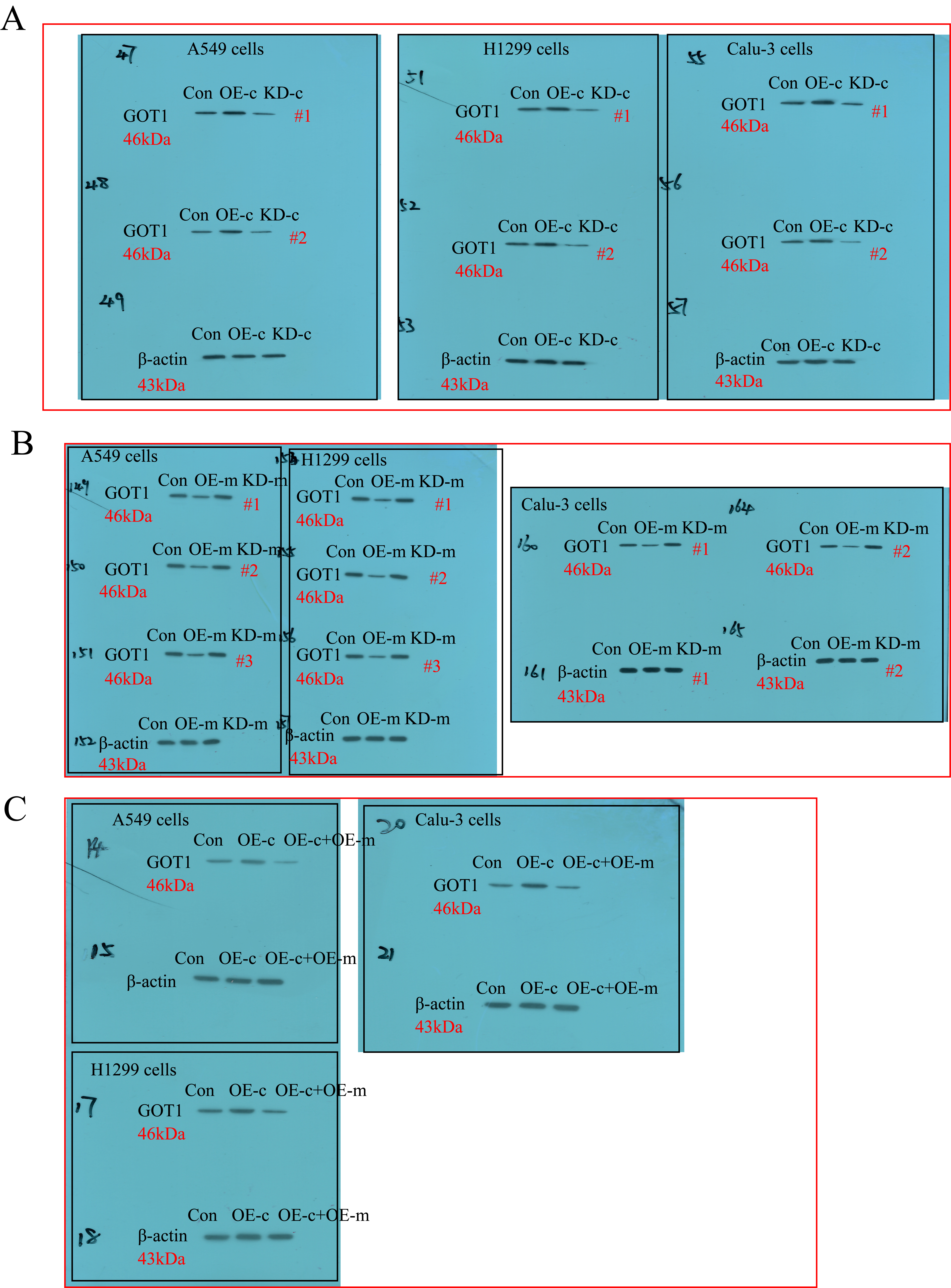
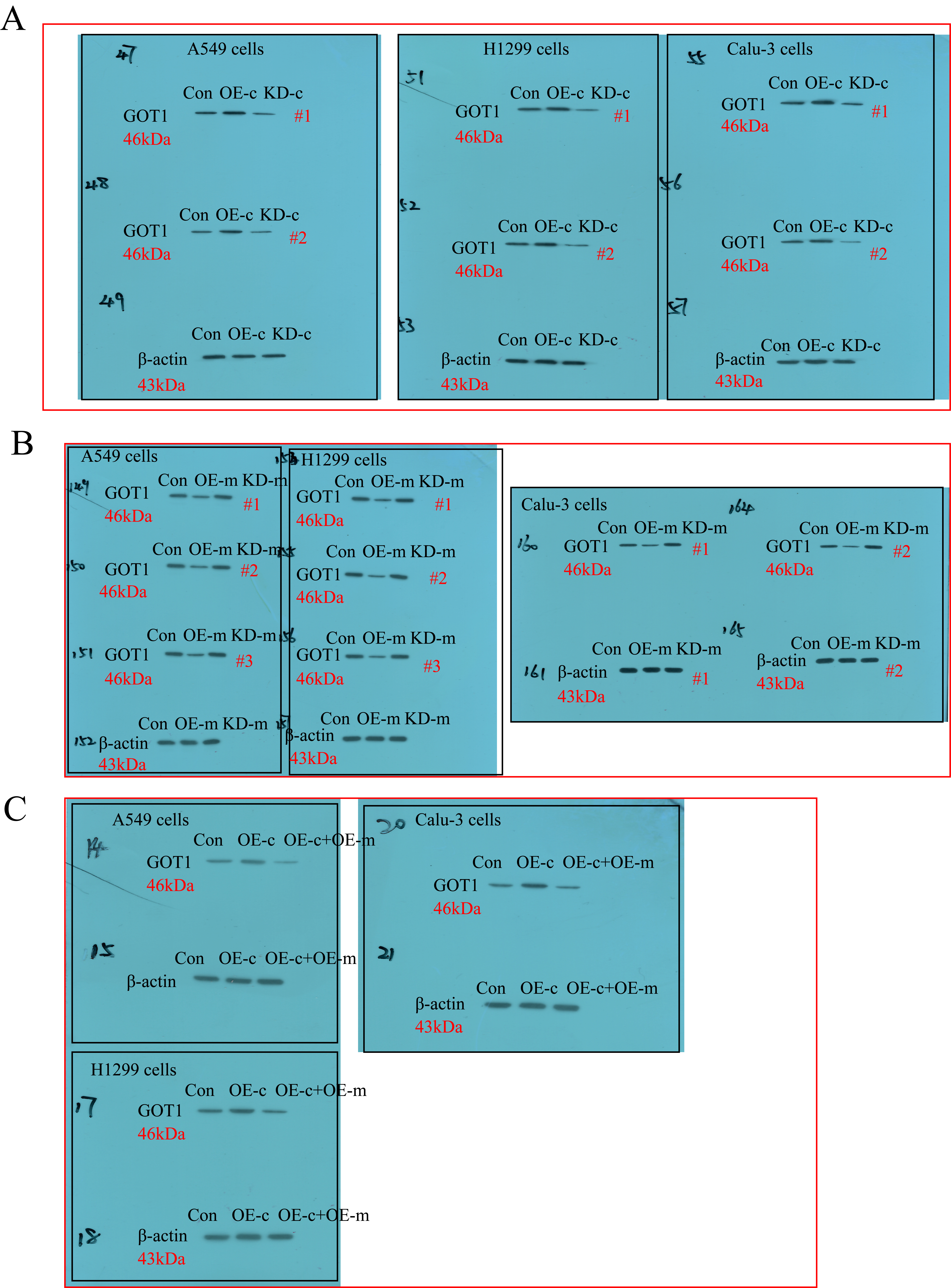
The overexpression and downregulation vectors for hsa_circRNA_103809 and GOT1, and miR-377-3p mimic and inhibitor were designed and synthesized by Sangon Biotech (Shanghai, China), and the above vectors were delivered into CS-NSCLC and CR-NSCLC cells to manipulate genes expressions by using the commercial Lipofectamine 2000 reagent (Invitrogen, USA), based on the experimental protocols provided by the producer. After that, Real-Time qPCR was conducted to validate the transfection efficiency of the above vectors. The sequence of siRNA for hsa_circRNA_103809 (5’- CAG TCT TAT CTC ACT TTA CTG GAT A-3’); The primers for hsa_circRNA_103809 overexpression plasmid construction (Forward: 5’-TAA TAA CTA AGA TCT GGT ACC GTT TTG ATG ATG AAA CAG AAG ATC AGC-3’, Reverse: 5’-GAA GCA TGA ATT CAA GGT ACC CAC CAA GTC TTC ACA ACT CCT GTC-3’); miR-377-3p mimic (5’-AUC ACA CAA AGG CAA CUU UUG U-3’) and inhibitor (5’-ACA AAA GUU GCC UUU GUG UGA U-3’); The short hairpin RNA (shRNA) for GOT1 downregulation (5’- CCG GGC GTT GGT ACA ATG GAA CAA ACT CGA GTT TGT TCC ATT GTA CCA ACG CTT TTT G-3’); The primers used for GOT1 overexpression (5’-CAA CTG GGA TTG ACC CAA CT-3’, Reverse: 5’-GGA ACA GAA ACC GGT GCT T-3’).
Cell counting kit-8 (CCK-8) assay
The NSCLC cells were pre-transfected with the above vectors, cultured in 96-well plates at standard culture conditions, and were subjected to cisplatin (25 μg/ml) stimulation for 0 h, 24 h, 48 h and 72 h, respectively. After that, the CCK-8 reaction solution (AbMole, USA) was incubated with the cells in the volume of 20 μl per well for 4 h at the incubator. Then, the plates were vortexed to thoroughly mix the cells with the solution, and were placed in a microplate reader (ThermoFisher Scientific, USA) to measure the optical density (OD) values at the wavelength of 450 nm, which could be used to represent relative cell proliferation in the cells.
Trypan blue staining assay
The CR-NSCLC and CS-NSCLC cells were pre-transfected with different vectors, and stimulated by using the cisplatin. Then, the cells were prepared and stained with trypan blue staining solution obtained from Invitrogen (USA) for 20 min at room temperature. After that, a light microscope was used to observe and count the number for dead blue cells, which were used to evaluate cell viability according to the following formula: Cell viability (%) = (Total cells – Dead blue cells)/Total cells * 100 %.
Annexin V-FITC/PI double staining assay
A apoptosis detection kit (Invitrogen, USA) was used to examine cell apoptosis in CS-NSCLC cells and CR-NSCLC cells, based on the protocols provided by the manufacturer. In brief, the cells were harvested and prepared, and subsequently stained with Annexin V-FITC and propidium iodide (PI) for 25 min at room temperature without light exposure. After that, a flow cytometer (FCM, ThermoFisher Scientific, USA) was used to examine the cell death ratio in NSCLC cells. Specifically, the early apoptotic cells were stained with Annexin V-FITC alone, the late apoptotic cells were stained with Annexin V-FITC and PI, and the necroptotic cells were stained with PI alone.
Real-Time qPCR
The NSCLC cells were subjected to differential treatments, and the TRIzol reagent (Invitrogen, USA) was employed to extract the total RNA. Specifically, 5 × 10 6 cells were treated with 1 ml TRIzol solution for 5 min, and were subsequently treated with chloroform for 15 min at room temperature. Next, the upper water phase was collected, and was treated with 0.5 ml isopropyl alcohol for 10 min. After centrifugation with 12,000 g for 10 min, 75 % ethyl alcohol was used to isolate and purify the total RNA. Next, the Real-Time qPCR was conducted to determine the expression levels of hsa_circRNA_103809, miR-377-3p and GOT1 mRNA, and the experimental procedures had all been documented in the previous publications [36, 37]. Of note, to detect hsa_circRNA_103809 levels, the total RNA must be pre-treated with RNase R enzyme (3 U/μg) for 20 min at 37 ℃ to eliminate linear RNA. The primer sequences for the involved genes are as follows: hsa_circRNA_103809 (Forward: 5’-ACG CAT TCT TCG AGA CCT CT-3’, Reverse: 5’-TGC CTG TAA CTC CTC TTC AGT-3’), miR-377-3p (Forward: 5’- ATC ACA CAA AGG CAA CTT TTG T-3’, Reverse: 5’- GGT GCA GGG TCC GAG GTA T-3’), GOT1 (Forward: 5’-TGC CAG TAG TGA AGA AAG TG-3’, Reverse: 5’-TAA GCG ATA GGA CCG AAT-3’), β-actin (Forward: 5’-GCT CGT CGT CGA CAA CGG CT-3’, Reverse: 5’-CAA ACA TGA TCT GGC TCA TCT TCT C-3’) and U6 (Forward: 5’-CTC GCT TCG GCA GCA CA-3’, Reverse: 5’-AAC GCT TCA CGA ATT TGC GT-3’).
Western Blot analysis
The RIPA lysis buffer was purchased from Solarbio (Beijing, China) to lyse the NSCLC cells/tissues and extract the total protein, according to the experimental procedures recorded in the previous publications [36, 37], the expression levels of GOT1, β-actin, cyclin D1, CDK2, cleaved caspase-3 and Bax were examined by using the Western Blot analysis. Specifically, the 40 μg/lane protein lysates were separated by using the 10 %-15 % SDS-PAGE, and the target protein bands were transferred onto the PVDF membranes (Millipore, USA). Next, the membranes were incubated with 5 % skim milk for 70 min at room temperature for blocking, and the membranes were probed with the primary antibodies against GOT1 (1:1500, MW: 50 kDa, #PA5-24634, Thermo, USA), β-actin (1:2000, MW: 42 kDa, #ab6276, Abcam, UK), cyclin D1 (1:1500, MW: 35 kDa, #ab40754, Abcam, UK), CDK2 (1:2000, MW: 33 kDa, #ab32147, Abcam, UK), cleaved caspase-3 (1:1500, MW: 17 kDa, #ab32042, Abcam, UK) and Bax (1:1500, MW: 21 kDa, #ab32503, Abcam, UK) overnight at 4 ℃. After washing by PBS buffer for 3 times, the PVDF membranes were incubated with the secondary antibody (Abcam, UK) for 120 min at room temperature. Finally, the protein bands were visualized by ECL system (GE Healthcare Bio-science, USA) and quantified by using the Image J software.
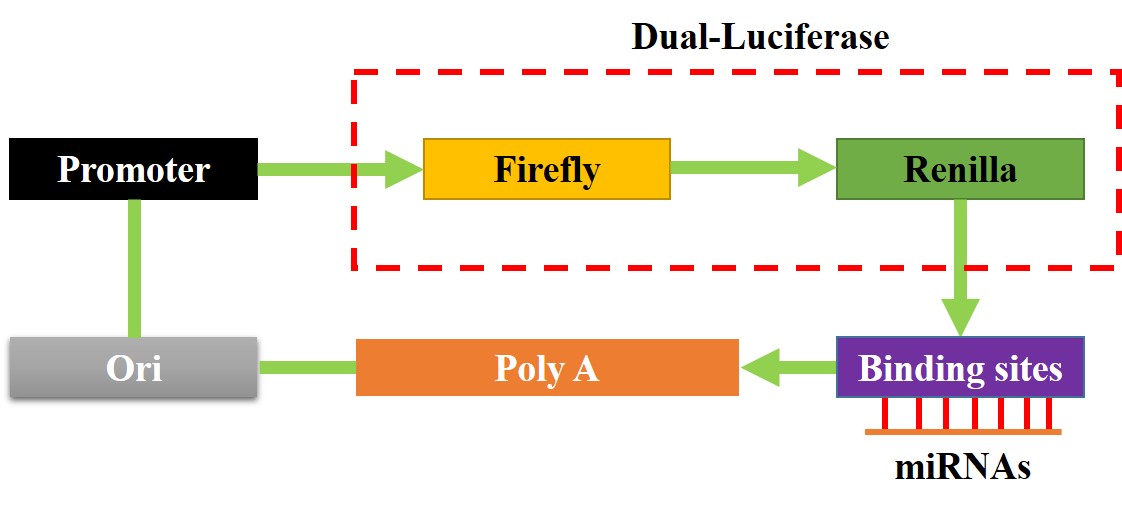
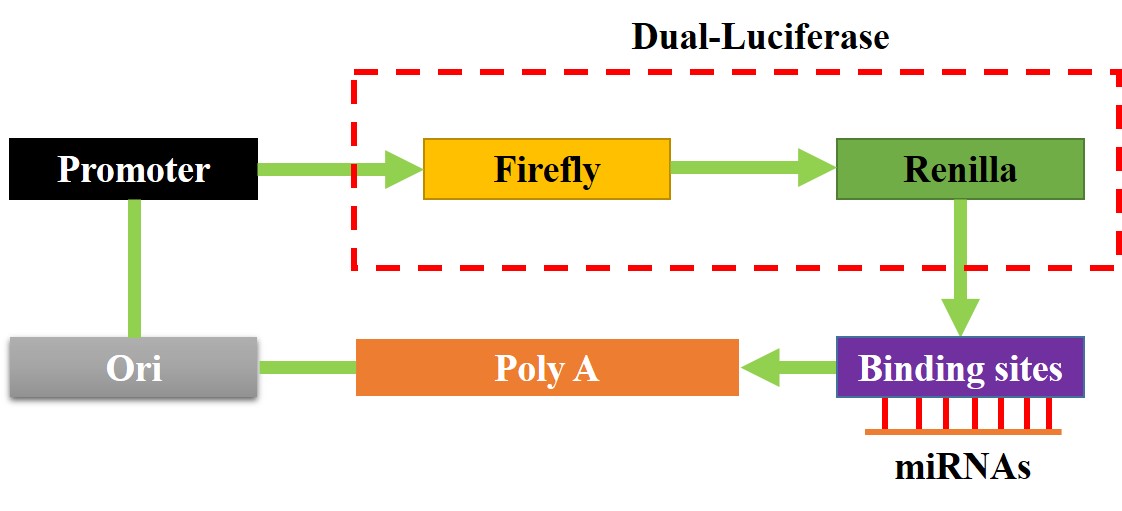
Dual-luciferase reporter gene system assay
The binding sites of miR-377-3p with hsa_circRNA_103809 and 3’ UTR region of GOT1 mRNA were predicted by the online miRDB software (http://mirdb.org/), and validated by using the dual-luciferase reporter gene system, and the detailed experimental procedures had been well documented in the previous literatures [36, 37]. Briefly, the targeting sites in hsa_circRNA_103809 and GOT1 were mutated, and named as Mut-circRNA and Mut-GOT1, respectively. Correspondingly, the original wild-type (Wt) genes were named as Wt-CircRNA and Wt-GOT1. The above sequences were cloned into the luciferase reporter vectors by Sangon Biotech (Shanghai, China), and the schematic image for the luciferase reporters was shown in Figure S3. The above vectors were delivered into NSCLC cells co-transfected with miR-377-3p mimic and inhibitor for 48 h. After that, the commercial dual-luciferase reporter assay kit (Promega, USA) was used to measure relative luciferase activities in the cells.
Xenograft tumor-bearing mice models
The CR-NSCLC cells were pre-transfected with different vectors, and were subcutaneously injected into the dorsal flank of male nude mice (N = 20), and the age of the mice ranged from 6 to 8 weeks. Each mouse was injected with 5 × 106 cells, at 7 days post-injection, the tumor were subjected to high-dose cisplatin (10 μg/ml) treatment every 3 days for 2 weeks. The above mice were equally divided into 4 groups, including Control, Cisplatin, KD-circRNA and Cisplatin + KD-circRNA, each group had 5 mice. The mice were sacrificed at 35 days post-injection. After that, the mice tumor tissues were collected, and the expression levels of proliferation associated proteins (cyclin D1 and CDK2) and apoptosis associated proteins (cleaved caspase-3 and Bax) were examined by using Western Blot analysis, and the expressions/localization of Ki67 protein in mice tissues were determined by Immunohistochemistry (IHC). All the animal experiments were approved by the Ethics Committee of Cancer Hospital of the University of Chinese Academy of Sciences, and the approval number was 2017DE453129832.
Immunohistochemistry (IHC)
The mice tumor tissues were collected and spliced into sections of 5 μm thickness, and IHC assay was conducted to determine the expressions and localization of Ki67 protein in the mice tissues, the detailed experimental procedures can be found at the previous publications [36, 37]. The antibody against Ki67 protein was bought from Abcam (UK), and was diluted at the ratio of 1:400.
Statistical analysis
Data analysis was conducted by using the SPSS 18.0 software, and the data was represented as Means ± Standard Deviation. The comparisons between two groups were performed by using the Student’s t-test, and the comparisons among multiple groups were conducted by using one-way ANOVA analysis. Each experiment was repeated at least 3 times, *P < 0.05 could be regarded as statistical significance.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}