All the required chemicals were purchased from Merck and Aldrich Chemical Company (USA). Precoated aluminium sheets (Silica gel 60 F254, Merck Germany) were used for thin-layer chromatography (TLC) and spots were visualized under UV light. Ct-DNA (as sodium salt) was obtained from SRL Pvt. Ltd, Mumbai, India. The concentrations of DNA were determined spectrometrically with an extinction coefficient of 6600 M− 1 cm− 1 at 258 nm. FT-IR spectra were recorded on Perkin Elmer model 1600 FT-IR RX1 spectrophotometer as KBr discs. 1HNMR was recorded on Bruker Spectrospin DPX 300 MHz BrukerSpectrospin using CDCl3 as a solvent and trimethylsilane (TMS) as an internal standard. Splitting patterns are designated as follows; s, singlet; d, doublet; m, multiplet. Chemical shift values are given in ppm. The ESI-MS was recorded on micrOTOF-Q II 10330 Electronspray ionization mass spectrometer (Bruker). X-ray data were collected on Bruker SMART Apex CCD diffractometer (SAI, Universidade da Coruña).
6.1 General procedure of synthesis of thiazolidinone derivative
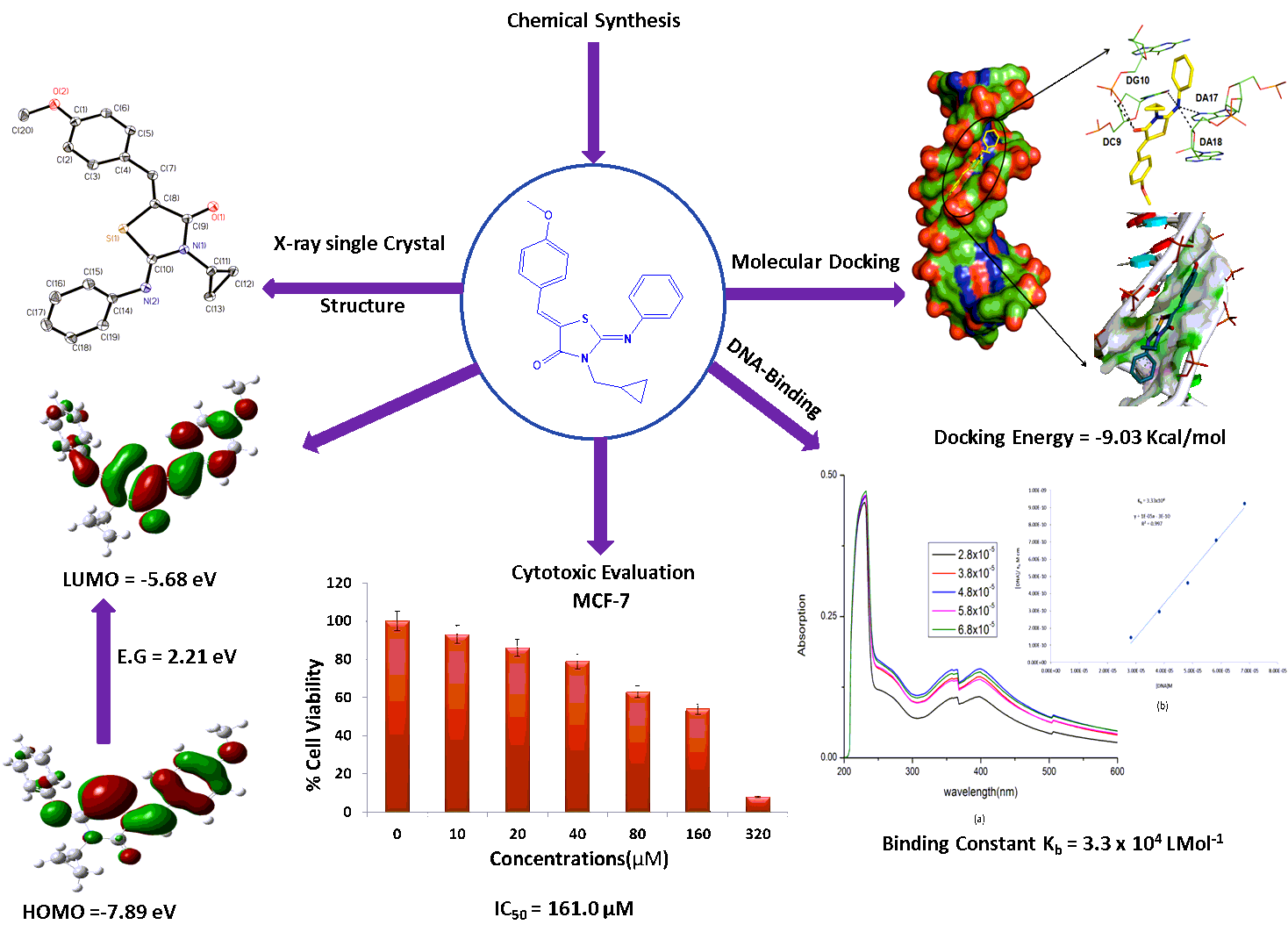
(1.0 mmol)- (2Z)-3-cyclopropyl-2-(phenylimino)-1,3-thiazolidin-4-one(4), was dissolved in absolute ethanol in a round bottom flask and (1mmol) p-methoxy benzaldehyde was added followed by the addition of (1.15 mmol) hexahydropyridine to the reaction mixture. The reaction mixture was refluxed for 11–12 h. The progress of the reaction was monitored by TLC. After the completion of reaction of the yellow precipitated solid appeared and collected by filtration, washed with ethanol. The obtained product was recrystallized in chloroform at room temperature. The shining yellow crystal was obtained.
6.1.1(2Z, 5Z)-3-cyclopropyl-5-[(4-methoxyphenyl)methylidene]-2-(phenylimino)-1,3-thiazolidin-4-one: Yield: 85%; IR (λmax) (cm− 1): 3013.21(Ar-H), 1710.14 (C = O), 1633.76(C = N),1592.87(C = C);1H-NMR (CDCl3)δ(ppm); 8.045 (s, 1H, H-C = C-), 7.391–7.280 (m, 3H, Ar-H), 7.171 (t, 1H, J = 4.2 Hz, Ar-H), 6.985 (d, 2H, J = 4.5 Hz, Ar-H), 6.933–6.830 (m, 3H, Ar-H),3.725(s, 3H, OCH3), 2.928–2.884 (m, 1H, cyclopropyl proton), 1.089–0.944 (m, 4H, cyclopropyl); 13C-NMR (CDCl3)δ(ppm);167.59 (C = O), 153.42 (-C = N-), 148.58(-C = C-), 152.82, 129.33, 125.92, 124.77, 121.12, 116.05, 114.66, 112.08(Aromatic), 56.06(OCH3), 25.92(-N-CH-), 7.97(cyclopropyl carbon-CH2); Calc. mass: 350.0; exp.mass: [M + H]+: 351.0 ; [M + 2H]+ : 352.0 ; [M + 3H]+: 353.0
6.2 Cytotoxicity studies (MTT assay)
Cell culture
Breast cancer cell line (MCF-7) was obtained from NCCS (Pune India). Cell was cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10 % fetal bovine serum (heat inactivated), 100 units/mL penicillin, 100 µg/mL streptomycin, and 2.5 µg/mL amphotericin B, at 37 0C in a relative humidity 80 %, 5 % CO2 [31].
MTT assay
The MTT assay is a standard colorimetric assay, in which mitochondrial activity is measured by splitting tetrazolium salts with mitochondrial dehydrogenases in viable cells only[32]. Cytotoxicity of compound (6) was evaluated through MTT (3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide, M2128 Sigma Aldrich) assay on MCF-7. MTT is a validated assay for the in vitro cytotoxicity of any natural, synthetic compounds and extracts. The cell count 1.2 × 104 cells/well were seeded in 96 well plate (150 µL/well). After the overnight incubation, cells were treated with different concentration of compound (6) for 48 h. After the 48 h of treatment, the medium was remove and incubated with 20 µL of MTT solution (5 mg/mL in Phosphate saline buffer) for 4 hour. The formazan crystals were formed by mitochondrial enzyme reduction, finally solubilized in DMSO (150 µL/well) and absorbance was recorded at 570 nm through the Microplate reader (iMark, BIORAD, S/N 10321). Percent viability was defined as the relative absorbance of treated versus untreated control cells.
6.3 DNA-binding
The stock solution of disodium salt of Ct-DNA was prepared in tris-HCl buffer (pH 7.2–7.3) and stored at 4 0C temperature. Once prepared, the stock solution was used within 4 days. The concentration of the solution was determined spectrometrically. The ratio of absorbance at 260 and 280 (≥ 1.8) indicated that DNA was sufficiently free of protein. The concentration of DNA was measured using its extinction coefficient at 260 nm (6600 M− 1 cm− 1) after dilutions. For the titration purpose, DNA stock solution was diluted using tris-HCl buffer. The compounds were dissolved in minimum amount of DMSO (1.6 x 10− 4M). UV-Vis absorption spectra were recorded after each addition of different concentrations of DNA. Absorption titration was conducted by adding varying concentrations (2.8–6.8 x 10− 5 M) of DNA. The intrinsic binding constant (Kb) was determined by Eq. (1), which was originally known as Benessi–Hilderbrand equation and further modified by Wolfe et. al.[33].
[DNA]/(ɛ a− ɛ f ) = [DNA]/(ɛ b− ɛ f ) + 1/K b (ɛb−ɛf) (1)
Where the apparent absorption coefficients ɛa, ɛf and ɛa correspond to A obs/[compounds], the extinction coefficient for the compounds, and the extinction coefficient for the compounds in the fully bound form. In plots of [DNA]/(ɛa−ɛf ) versus [DNA], Kb is given in the ratio of the slope to intercept.
6.3 DNA docking studies
Docking studies were performed at Intel(R) Core(TM) i3 CPU (2.3 GHz) with XP-based operating system (Windows 2010). 3D Structures of the compounds (6) was drawn by Marvin sketch and saved in pdb file format. The preparation of the compound was done by assigning Gastegier charges, merging non-polar hydrogens, and saving it in PDBQT file format using Auto-Dock Tools (ADT4.2)[29, 34, 35]. The X-ray crystal structure of DNA (PDB ID: 1BNA) was obtained from the Protein Data Bank [http://www.rcsb.org/pdb]. Using ADT 4.2, DNA was saved in PDB file format leaving heteroatoms (water). Gastegier charges were assigned to DNA and saved in PDBQT file format. Preparation of parameter files for grid and docking was done using ADT. Docking was performed with Auto Dock Vina 4.2[36] considering all the rotatable bonds of ligand (compound 3 and 4) as rotatable and receptor (DNA) as rigid[37]. A grid box of size 64 × 64 × 118 Å with 0.375Å spacing was used that included the whole DNA. The final structure of the docked complexes was drawn using PyMol[38] and 2D plot of docked complexes were constructed using Schrödinger visualizer (Maestro 10.5 trial version, Maestro, 2016).
6.4 X- Ray crystal structure determination
Three-dimensional X-ray data were collected on a Bruker Kappa Apex CCD diffractometer at low temperature by the φ-ωscan method. Reflections were measured from a hemisphere of data collected from frames, each of them covering 0.3º in ω.A total of 59758 for 6, reflections measured were corrected for Lorentz and polarization effects and for absorption by multi-scan methods based on symmetry-equivalent and repeated reflections. Of the total, 4744 independent reflections exceeded the significance level (F/σF) > 4.0. After data collection, an multi-scan absorption correction (SADABS)[39] was applied, and the structure was solved by direct methods and refined by full matrix least-squares on F 2 data using SHELX suite of programs [40]. Hydrogen atomswerelocated in difference Fourier map and left to refine freely, except for C(20), which were included in calculation position and refined in the riding mode. Refinements were done with allowance for thermal anisotropy of all non-hydrogen atoms. A final difference Fourier map showed no residual density outside: 0.403 and − 0.265 e.Å−3.A weighting scheme w = 1/[σ2(Fo 2) + (0.052700 P)2 + 0.584300P] for 6, where P = (|Fo|2 + 2|Fc|2)/3, was used in the latter stages of refinement. Further details of the crystal structure determination are given in Table (3). CCDC 2006786 contains the supplementary crystallographic data for the structure reported in this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html , or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+ 44) 1223 336 033; or e-mail: [email protected]. Supplementary data associated with this article can be found, in the online version, at doi: $$$$$.
Table (3): Crystal Data and Structure Refinement for the compound (2Z,5Z)-3-cyclopropyl-5-[(4-methoxyphenyl)methylidene]-2-(phenylimino)-1,3-thiazolidin-4-one(6)
|
Compound
|
6
|
|
Formula
|
C20 H18 N2 O2 S
|
|
Formula weight
|
350.42
|
|
T, K
|
100(2)
|
|
Wavelength, Å
|
0.71073
|
|
Crystal system
|
Monoclinic
|
|
Space group
|
P21/n
|
|
a/Å
|
11.9703(6)
|
|
b/Å
|
7.0182(4)
|
|
c/Å
|
20.4169(11)
|
|
β/º
|
93.083(2)
|
|
V/Å3
|
1712.74(16)
|
|
Z
|
4
|
|
F000
|
736
|
|
Dcalc/g cm− 3
|
1.359
|
|
µ/mm− 1
|
0.205
|
|
θ/ (º)
|
3.37 to 31.64
|
|
Rint
|
0.0399
|
|
Crystal size/ mm3
|
0.50 x 0.48 x 0.32
|
|
Goodness-of-fit on F2
|
1.038
|
|
R1[I > 2σ(I)] a
|
0.0356
|
|
wR2 (all data) b
|
0.1001
|
|
Largest differences peak and hole (eÅ−3)
|
0.403 and − 0.265
|
aR1 = S½çFo÷ - êFc÷½/S½Fo½. bwR2 = {S[w(½çFo÷2 -êFc÷2½)2]½/S[w(Fo2)2]}1/2

{kind=link}