Impact of light perception impairment on the fruit transcriptome
To investigate the role played by either PHYA or PHYB1/PHYB2 (hereafter PHYB1B2) in overall gene expression during fruit development, the transcriptome of fruits at the immature green (IG) and breaker (BK) stages from phyA and phyB1B2 null mutants as well as their wild-type (WT) counterpart, was determined by RNAsEq. Among the approximately 20,000 transcriptionally active loci in each biological replicate (Supplementary Table 1), 1.2% and 2.4% at the IG stage and 9.1% and 11.2% at the BK stage were identified as differentially expressed genes (DEGs) in phyA or phyB1B2 mutants, respectively, compared to the WT (Fig. 1a; Supplementary Table 2). For both genotypes, the number of exclusive DEGs was significantly lower in the IG stage than in the BK stage; similarly, the number of genes that were commonly regulated by PHYA and PHYB1B2 was 172 at the IG stage and 785 at the BK stage (Fig. 1b). Subsequently, the altered expression of approximately 76% (23/30) of the tested genes was validated by RT-qPCR (Supplementary Table 3). Comparison with previously reported expression data for genes involved in ripening regulation, ethylene biosynthesis and signalling, and carotenogenesis further validated our RNAseq data, as 90% of the analysed genes on average showed the expected mRNA profile at IG and BK stages. It is worth mentioning that most of the genes displayed the same transcript fluctuation in the WT, phyA and phyB1B2 genotypes, though this was somewhat attenuated in the mutants (Supplementary Table 4). These results showed that PHY-meditated light perception regulates more genes in BK than in the early stages of fruit development and that PHYB1B2 has a more substantial impact than PHYA in the fruit transcriptome in both analysed stages.
A closer look at DEGs function revealed a similar distribution of loci across MapMan categories in response to phyB1B2 and phyA mutations in both developmental stages, although with distinct abundance levels (Fig. 1c). At the IG stage, eight categories were mainly represented, including at least 2% of the DEGs identified in phyA and phyB1B2: photosynthesis, lipid metabolism, phytohormone action, RNA biosynthesis, protein modification, protein homeostasis, cell wall organization, and solute transport (Fig. 1c; Supplementary Tables 5 and 6). It is worth highlighting the abundance of the DEGs within the photosynthesis category in the phyB1B2 mutant, among which 34 out of the 37 genes were downregulated (Supplementary Table 6). In the BK stage, at least 2% of the DEGs were related to the lipid metabolism, phytohormone action, RNA biosynthesis, protein modification and homeostasis, cell wall organization and solute transport categories in both genotypes (Fig. 1c; Supplementary Tables 7 and 8). However, while phyA deficiency also affected carbohydrate metabolism and external stimuli (Supplementary Table 7), the phyB1B2 mutant showed a large number of DEGs related to the cell cycle and chromatin organization (Supplementary Table 8). Interestingly, the chromatin organization category displayed 52 DEGs, 45 of which were upregulated. These genes encode nucleosome constituent histones (H3, H4, H2A and H2B); DNA methylases/demethylases; histone post-translational modifiers such as deacetylases, methylases/demethylases, histone ubiquitination factors and histone chaperones; chromatin remodelling factors; and genes involved in RNA-independent and RNA-directed DNA methylation (Supplementary Table 8). These results led us to further investigate the impact of DNA methylation on PHY-mediated gene expression reprogramming.
PHYs regulate the epigenome profile
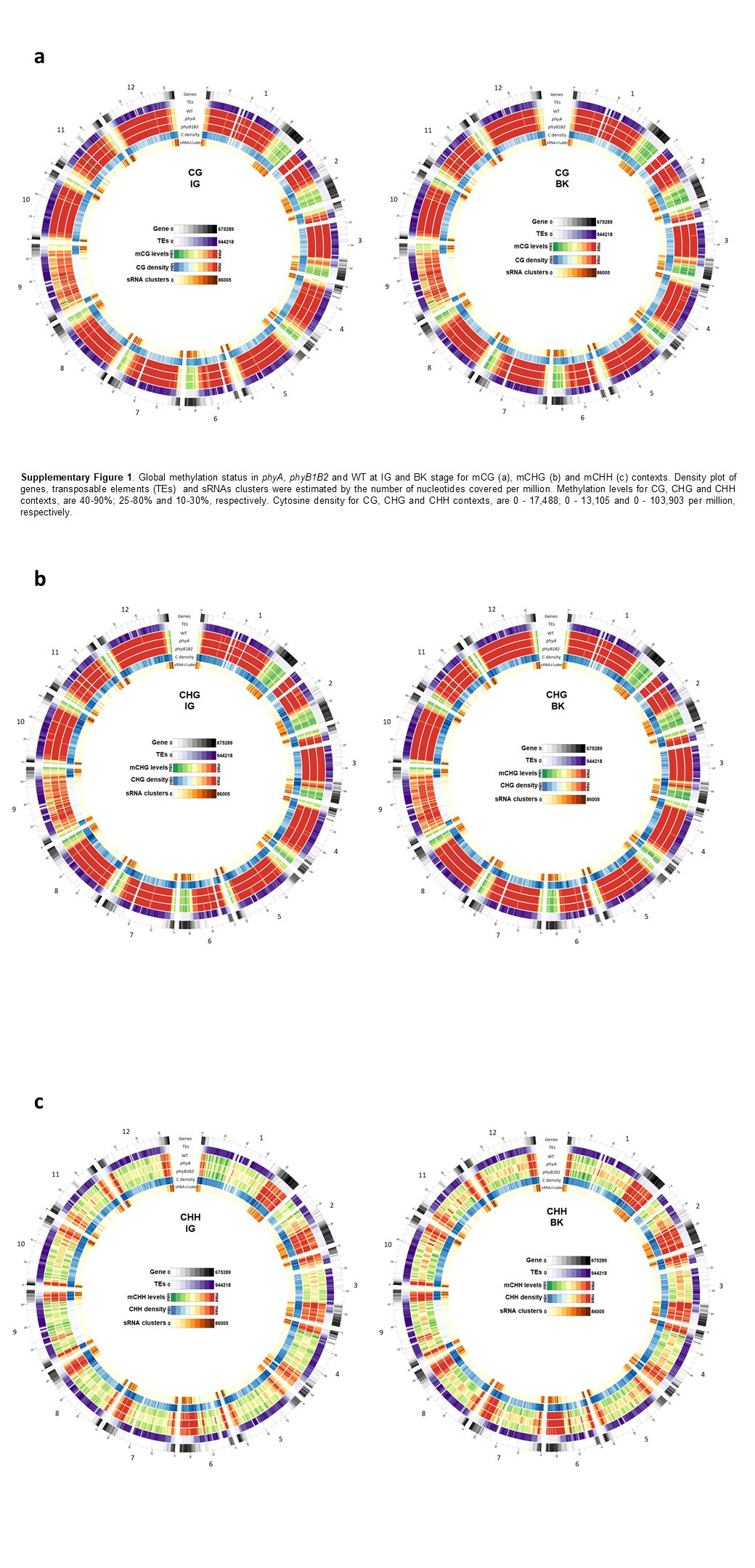
The global profile of methylated cytosines (mCs) in the epigenome of tomato fruits was assessed by whole-genome bisulfite sequencing in the IG and BK stages for phyA, phyB1B2 and WT genotypes. In agreement with previous reports27,28, regardless of the genotype and fruit stage, the greatest total number of mCs was located in the CHH context, followed by the CG and CHG contexts, while the methylation level was highest in the CG (80%) context followed by the CHG (67%) and CHH (23%) contexts (Supplementary Table 9, Supplementary Fig. 1). For further comparisons, we selected only cytosines with coverage > 10X, and except for chromosome 9 in the transposable elements TEs enriched region, all samples met this cutoff. In all contexts, the highest cytosine density was associated with gene-rich euchromatic regions located at chromosome arm ends (Supplementary Fig. 1). Conversely, in symmetrical contexts (CG and CHG), the highest methylation rates were found across pericentromeric regions enriched in TEs and in the CHH context in gene-rich regions associated with a higher density of sRNAs (Supplementary Fig. 1). The comparison of the methylation status between the two fruit stages showed that ripening-associated demethylation27 occurs mainly in the CG context, especially in gene-rich regions, and that it is impaired in phyB1B2 mutant BK fruits (Supplementary Fig. 1).
The subsequent comparison between genotypes revealed global epigenome alteration in phy mutants in all contexts analysed. The most remarkable observation was the presence of considerable hypermethylation in all contexts across gene-rich regions in BK-stage fruits from phyB1B2 (Fig. 2a). In contrast, phyA exhibited hypermethylation in CHG and CHH contexts associated with TE-rich regions (Fig. 2a), suggesting that different PHYs control DNA methylation across specific genomic regions through distinct regulatory mechanisms. Interestingly, PHY-associated hypomethylation was exclusively detected in the CG context of gene-rich regions in IG-stage fruits from phyA and in the CHH context of TE-rich regions for BK-stage fruits from phyB1B2. In summary, these data revealed that both PHYA and PHYB1B2 regulate the global methylome, but PHYB1B2 has a greater impact on ripening-associated methylation reprogramming across gene-rich genomic regions in tomato fruits.
To investigate the relationship between PHY-regulated cytosine methylation and gene expression, we first identified genes with differentially methylated promoters (DMPs, 2 kb upstream of the transcription start site) in all three contexts. Interestingly, associated with the massive alteration previously observed, the pattern of DMPs varied with the mC context, stage and genotype (Fig. 2b, Supplementary Tables 10 and 11). Regarding the CG context, whereas the phyA mutant showed virtually the same frequency of hyper- and hypomethylated promoters in the two stages, the status of hypermethylated promoters in phyB1B2 increased over 60% from the IG to BK stage, while the number of loci with hypomethylation decreased 50% (Fig. 2b, Supplementary Table 12). In contrast, phyA showed a greater number of hypermethylated promoters in the CHG context in the IG stage than in the BK stage, while the levels in the WT and phyB1B2 mutant remained similar upon ripening (Fig. 2b, Supplementary Table 13). In the CHH context, the number of hypermethylated promoters decreased in both genotypes from the IG to BK stages (Fig. 2b, Supplementary Table 14).
These results indicate that PHY deficiency results in massive promoter hypermethylation in both the IG and BK stages of tomato fruit development. Moreover, they reinforce the role of PHYB1B2 in ripening-associated demethylation and its putative effect on gene expression.
Effect of PHY-mediated differential methylation on the transcriptome
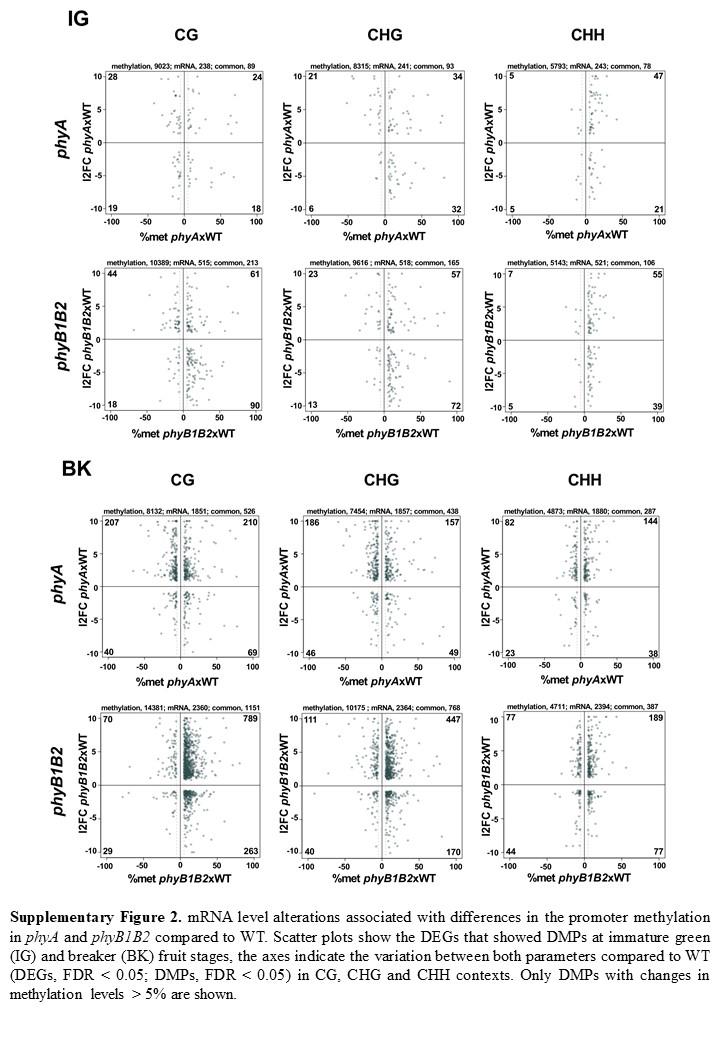
To assess whether the differential methylation of gene promoters affects mRNA levels, we crossed data from DEGs and DMPs between genotypes. Supplementary Fig. 2 shows scatter plots of promoter methylation vs mRNA fold changes for comparisons of the two genotypes at the two examined developmental stages in the three mC contexts. The most evident result was that among the thousands of loci with identified DMPs (Fig. 2b), only hundreds of the loci were also differentially expressed (Supplementary Table 15) (0.7% for IG phyA, 1.6% for IG phyB1B2, 5.6% for BK phyA and 7.4% for BK phyB1B2), raising an intriguing question about the biological significance of the extensive change in the methylation pattern observed in the mutants. In contrast, the percentages of the DEGs showing DMPs were 73% for IG phyA, 76% for IG phyB1B2, 72% for BK phyA and 75% for BK phyB1B2 (Supplementary Fig. 2). Many more DEGs with DMPs were observed in BK than in IG fruits and in phyB1B2 than in the phyA genotype. The functional categorization of these genes revealed a similar category distribution to the DEGs (Fig. 1c, Supplementary Table 16–19). At the IG stage, there were seven categories in which at least 2% of the loci showed DMPs and differential expression in both genotypes: photosynthesis, phytohormone action, RNA biosynthesis, protein modification and homeostasis, cell wall organization and solute transport, whereas phyB1B2 additionally impacted lipid metabolism (Fig. 1c). In the BK stage, the categories in which at least 2% of the DEGs showed DMPs were lipid metabolism, phytohormone action, RNA biosynthesis, protein modification and homeostasis, cell wall organization and solute transport-related functions in both genotypes, while only phyA impacted carbohydrate metabolism and external stimuli, and only phyB1B2 affected photosynthesis, chromatin organization and cell cycle categories.
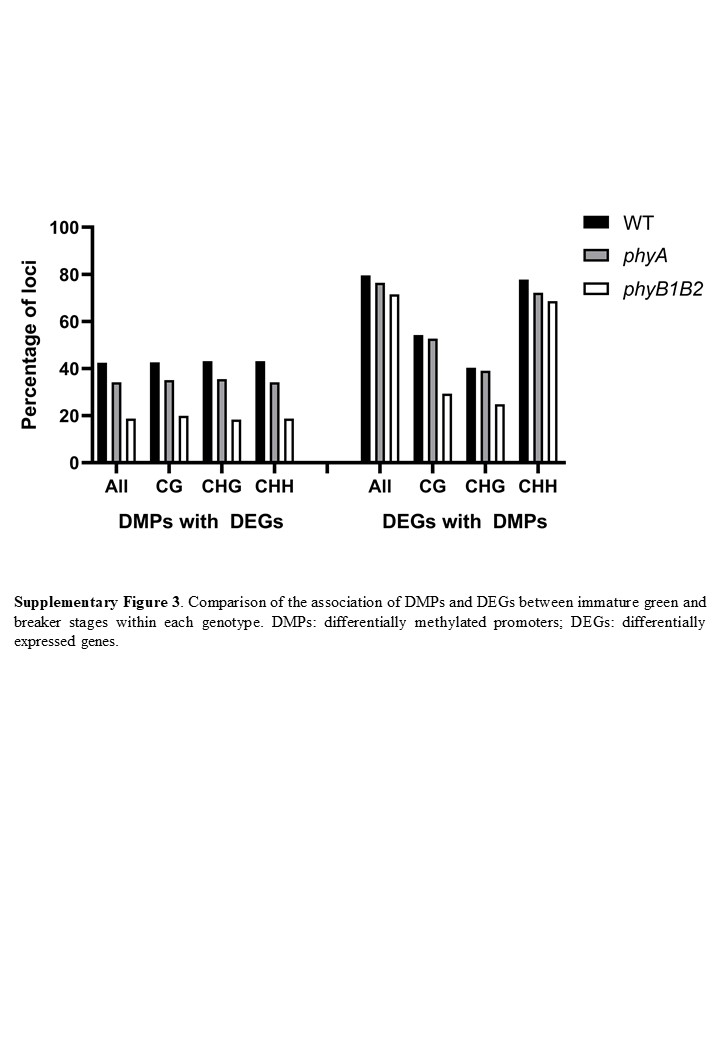
Interestingly, in the comparison of the IG and BK stages, 42.5%, 34.2% and 18.8% of the DMPs were associated with DEGs, while 79.5%, 76.6% and 71.5% of the DEGs showed differences in promoter methylation in WT, phyA and phyB1B2, respectively (Supplementary Fig. 3). All of these data demonstrated that the PHY-dependent mRNA profile is profoundly affected by promoter methylation; however, massive genome-wide PHY-induced methylation reprogramming has a still uncharacterized role beyond the regulation of mRNA accumulation. Moreover, promoter methylation has a greater effect on gene expression regulation during BK than in the IG stage. Additionally, the data showed that PHYB1B2 has a more extensive influence on gene expression regulated via promoter methylation than PHYA, reinforcing the above conclusion that PHYB1B2 affects CG ripening-associated demethylation (Supplementary Fig. 2).
The sRNAome is altered by PHY deficiency
To assess the involvement of RdDM in PHY-mediated transcriptome regulation, the sRNAome was analysed in fruits at the IG and BK stages from both mutants and the WT genotype (Supplementary Table 20a). A total of 28,314 clusters of sRNAs were identified across the whole genome in at least one of the samples, including 7,984 in gene bodies, 7,863 in promoter regions, 7,966 in TEs and the remaining 4,501 across intergenic regions (Supplementary Fig. 1, Supplementary Table 20b). The methylation level was evaluated for each sRNA cluster-targeted genomic region (sCTGR), and as previously observed for promoter regions, a higher proportion of hypermethylation was observed in BK fruits from phyB1B2 in the CG symmetrical context. Moreover, the greatest number of differentially methylated sCTGRs was observed in the asymmetrical context CHH. (Fig. 3a, Supplementary Table 20g-j).
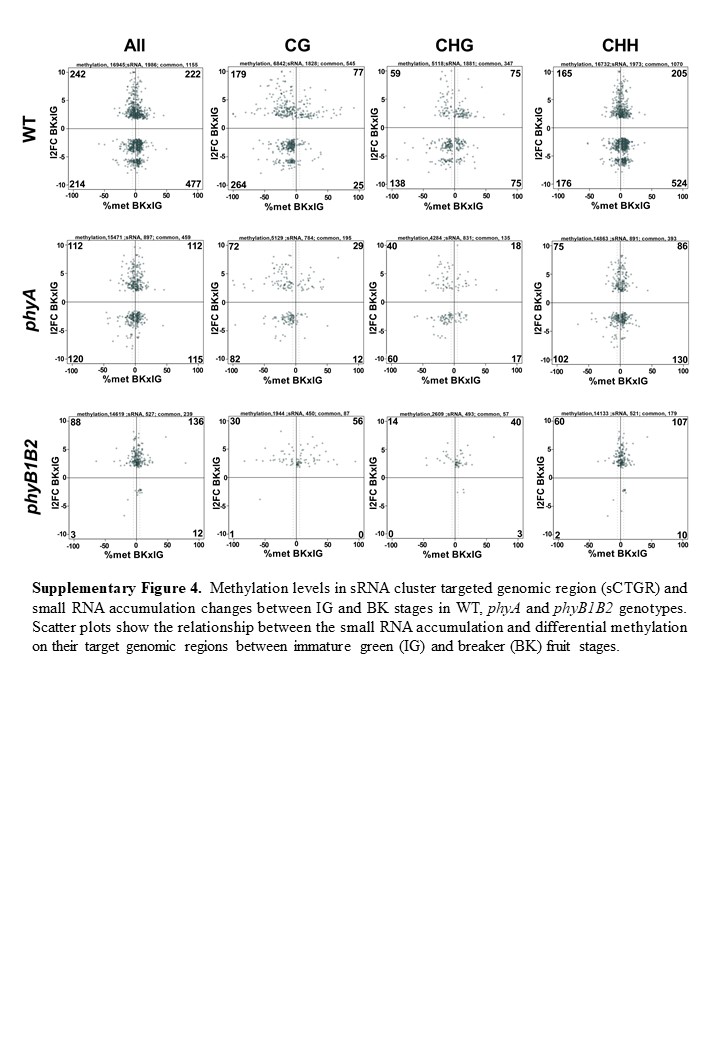
sCTGR methylation levels and sRNA accumulation data were intersected, and among a total of 154, 318, 267 and 257 differentially accumulated sRNA clusters (Supplementary Table 20c-f), 88, 154, 99 and 82 also showed differential methylation changes (> 5%) in phyA IG, phyB1B2 IG, phyA BK and phyB1B2 BK fruits, respectively (Fig. 3b, (Supplementary Table 20g-j), showing a strong association (P < 0.005) of the two datasets. Intriguingly, this positive association was not observed in the transition from the IG to BK stages (Supplementary Fig. 4), suggesting that the global methylation changes via RdDM could be attributed to PHY deficiency. Moreover, a clear disturbance in sRNA accumulation was observed in phyB1B2, since almost no clusters with less sRNA accumulation were observed in BK compared to the IG stage (Supplementary Fig. 4). We further analysed whether this positive correlation observed between sRNA accumulation and sCTGR methylation impacted gene expression levels. Notably, regardless of the fruit developmental stage, changes in the accumulation of sRNA located in gene bodies (GBs), and not in the promoter (P) region, were positively correlated with the mRNA level (Fig. 3c, Supplementary Table 20k-n). Among these loci, two interesting examples were identified: the well-known ripening-associated genes RIPENING INHIBITOR (RIN, Solyc05g01202029) and FRUITFULL2 (FUL2, Solyc03g11483030), which showed higher expression in phyB1B2 at the IG stage (Fig. 4a) and higher sRNA accumulation and sCTGR methylation across their GBs (Fig. 4b) compared to WT. The premature expression of these TFs was in agreement with the previously reported anticipation of ripening onset in the phyB1B2 mutant31. Altogether, these findings revealed (i) impaired RdDM in BK fruits of phyB1B2, indicated by the absence of clusters with less sRNA accumulation, and (ii) that GB RdDM is an important mechanism that positively regulates gene expression in a PHY-mediated manner during fruit development.
PHYB1B2-dependent methylation regulates fruit chlorophyll accumulation
The categorization of DEGs associated with differential promoter methylation revealed prominent representation of the photosynthesis category in the fruits of the phyB1B2 mutant at the IG stage (Fig. 1c). Among the 32 genes, 22 were downregulated and hypermethylated in the promoter region (Supplementary Tables 6 and 17). Most of these genes encode chlorophyll-binding proteins, structural photosystem proteins and chlorophyll biosynthetic enzymes. This might at least partly explain the reduction of 50% in the total chlorophyll level observed in phyB1B2 IG fruits (Fig. 5a). Detailed promoter analysis of the chlorophyll-related enzymes PROTOCHLOROPHYLLIDE OXIDOREDUCTASE 3 (POR3, Solyc07g054210), and two CHLOROPHYLL A/B BINDING PROTEINs (CBP, Solyc02g070990 and CAB-3c, Solyc03g005780) showed that the reduced mRNA levels of these three genes in phyB1B2 (Fig. 5b) were correlated with the presence of hypermethylated regions in the promoters. Interestingly, several of these hypermethylated sequences overlapped with HY5 and PIF photomorphogenic TF binding sites, such as G-box (CACGTG), CA hybrid (GACGTA), CG hybrid (GACGTG) and PBE-box (CACATG) motifs32 (Fig. 5c). These results suggest that the transcription of genes involved in chlorophyll metabolism and the photosynthetic machinery in tomato fruits is regulated by the PHYB1B2-mediated methylation status of their promoter regions in addition to the PHY-mediated post-translational regulation of HY5 and PIF protein levels2.
The methylation-mediated regulation of fruit ripening is PHYB1B2-dependent
In their seminal study, Zhong et al. (2013)27 revealed that the extensive methylation in the promoter regions of ripening-associated genes gradually decreases during fruit development. Interestingly, RNA biosynthesis, which includes transcription factors, was the most abundant functional category among the DEGs that showed DMPs (Fig. 1c). Thus, we examined a set of ripening-associated master transcription factors: RIN, Solyc05g012020, NON-RIPENING (NOR, Solyc10g00688033), COLORLESS NORIPENING (CNR, Solyc02g07792034) and APETALA2a (AP2a, Solyc03g04430035). The evaluation of the promoter regions clearly showed that while their methylation level decreased from the IG to BK stage in the WT genotype, they remained highly methylated in phyB1B2 (Fig. 6a). As previously observed, several hypermethylated regions are closely linked to HY5 and PIF binding sites. The maintenance of high methylation levels in the promoters of these key regulatory genes at the onset of fruit ripening was highly correlated with their transcriptional downregulation at the BK stage (Fig. 6b).
Carotenoid accumulation is probably the most appealing and best investigated trait of tomato fruits; in agreement with previous findings26, ripe phyB1B2 fruits showed a five-fold reduction in carotenoid content compared to WT (Fig. 7a).
With the aim of evaluating whether this effect is a consequence of the methylation-mediated regulation of carotenoid biosynthesis genes, we further analysed the promoters of PHYTOENE SYNTHASE 1 (PSY1, Solyc03g031860), PHYTOENE DESATURASE (PDS, Solyc03g123760), 15-CIS- ζ-CAROTENE (ZISO, Solyc12g098710) and ZETA-CAROTENE DESATURASE (ZDS, Solyc01g097810), which, with the exception of PDS, were hypermethylated in phyB1B2 BK fruits (Supplementary Table 11). The mC profile confirmed the presence of hypermethylated regions in all four promoters, which were predominantly located near light-dependent TF binding motifs in the PSY1, PDS and ZISO promoters (Fig. 7b), explaining the reduced mRNA levels of these genes observed in phyB1B2 (Fig. 7c).
RIN is one of the main TFs controlling ripening-associated genes by directly binding to their promoters. RIN binding occurs in concert with the demethylation of its targets27. To examine whether RIN binding site methylation could be affected by the phyB1B2 mutation in the ripening-related master transcription factors and carotenoid biosynthesis gene promoters, we mapped the available RIN ChIP-seq data27 and performed de novo motif discovery (Supplementary Fig. 5). Interestingly, the levels of mCs associated with RIN targets were higher in the NOR, CNR and AP2a promoters in phyB1B2 than in WT. Moreover, the RIN promoter itself was hypermethylated across the RIN binding site in phyB1B2 BK fruits, suggesting a positive feedback regulatory mechanism (Fig. 6a). Finally, in the phyB1B2 mutant, the PSY1, PDS, ZISO and ZDS promoters showed higher methylation overlapping with RIN target binding motifs, indicating that the upregulation of carotenoid biosynthesis genes during tomato ripening is dependent on the PHYB1B2-mediated demethylation of RIN target sites.
Altogether, our findings showed that PHYB1B2 is a major player in fruit ripening by controlling the promoter demethylation of master transcriptional regulators and carotenoid biosynthesis genes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}