2.1 Rat model of crushed sciatic nerves (CSN)
All experimental protocols of animal model were approved by the Ethics Committee of the Shanghai General Hospital, Shanghai JiaoTong University School of Medicine. Healthy Sprague Dawley (SD) rats (180-220g) were obtained from the Zhejiang Chinese Medical University Laboratory Animal Research Center (Zhejiang, China) and maintained in 12-h-light/12-h-dark conditions with ad libitum access to food and water. Every effort was made to minimize the number of animals used and their suffering. The rat model of CSN was carried out according to the previous description (24). In brief, all rats were anesthetized with 1% pentobarbital sodium (40 mg/kg), fixed on the operating table and disinfected with 1% iodophor solution. The upper right femoral posterior incision was taken, skin and subcutaneous fascia were incised layer by layer, and the sciatic nerve was fully exposed. At a distance of 0.5 cm from its bifurcation, the nerve was compressed with a pair of 14-cm hemostat forceps for 30 sec.
2.2 RNA-sequencing
Total RNA was extracted from the distal stumps of CSN (0.5 cm) and intact contralateral nerves at 4 dpi as described previously (25). In brief, RNA was treated with amplification-grade DNase Ι (Invitrogen, Carlsbad, CA, USA). To quantify circRNAs, Epicentre RNaseR (Lucigen, Middleton, WI, USA) was applied to degrade linear RNAs and subsequently purified the resulting using a RNeasy MinElute Cleanup Kit (Qiagen, Duesseldorf, Germany). Total RNA of each sample was quantified using the NanoDrop 1000 Spectrophotometer (Nanodrop, Waltham, MA, USA). The total RNA samples (3 mg) were treated with the RiboMinus Eukaryote Kit (Invitrogen) to remove rRNA. cDNA libraries were generated as the Illumina TruSeq RNA Sequencing (RNA-Seq) protocol and sequenced on an Illumina HiSeq 2000 sequencing platform.
2.3 Electrophysiological Assessment
At 28 days post-injury (dpi), rats were subjected to an electrophysiological test according to previously described protocols (n = 5) (26). In brief, the sciatic nerve near the repair site was re-exposed, a pair of stimulating electrodes (13 mm long, 0.5 mm in diameter) was inserted 3 mm near the crushed site to stimulate the sciatic nerve, and a pair of needle electrodes was inserted subcutaneously into the middle of the intrinsic foot muscle to record the compound muscle action potential (CMAP) using EMG Evoked Potentiometer (MEB-9200K, Nihon Kohden, Japan). The amplitude and latency of each test were analyzed to determine the nerve conduction intensity and nerve conduction velocity, respectively.
2.4 Walking track analysis
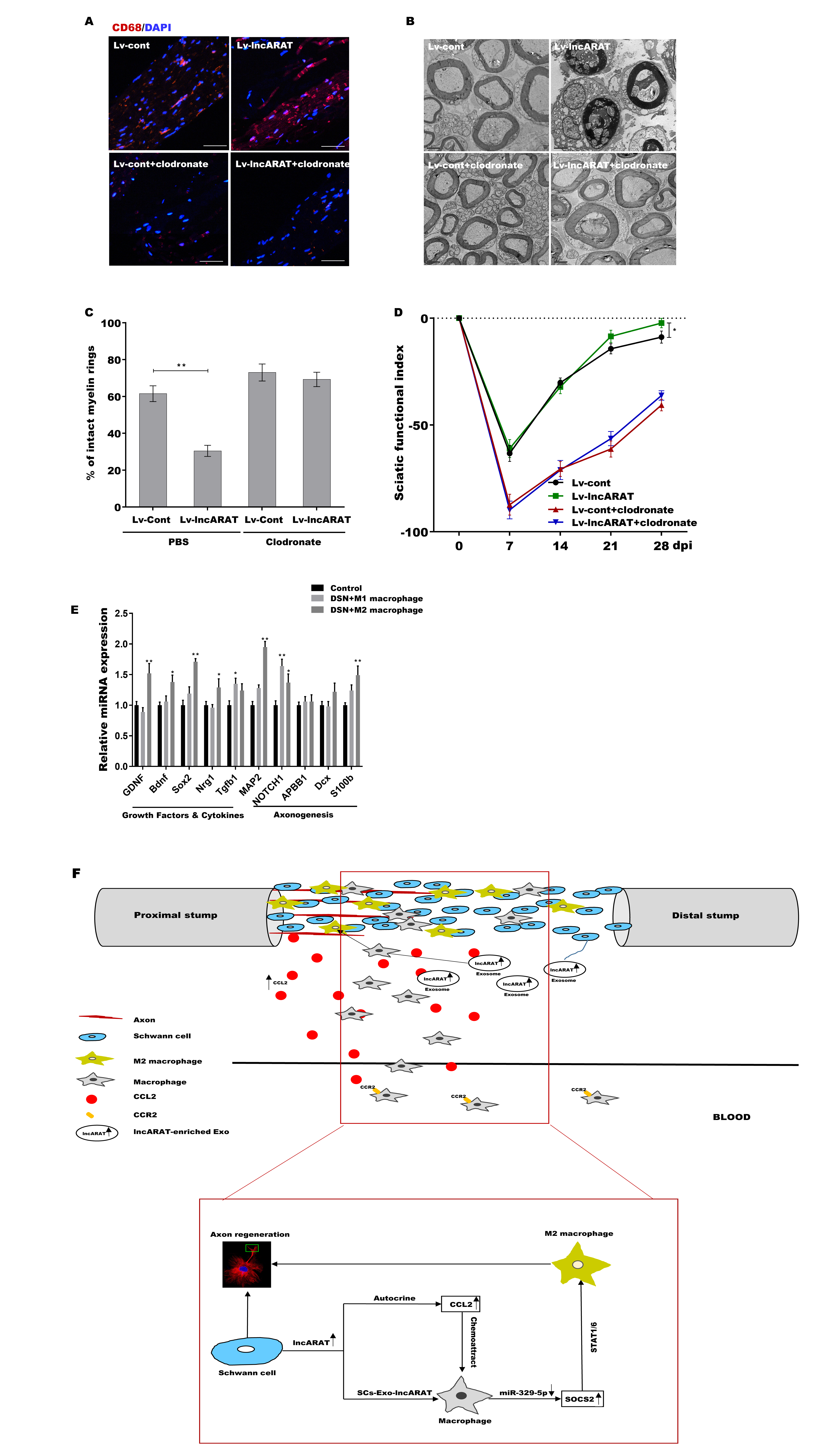
To perform functional analysis of movement, we applied propylene pigments to the plantar surface of the hind paws of rats (28 dpi) and allowed them to walk along white paper-covered corridors. Footprints from ipsi- and contralateral paws were analyzed through measuring the print length (PL) and the distance between 1st and 5th toes. The three parameters were combined to obtain the Sciatic Functional Index (SFI) (27), which quantifies the changes in the walking pattern (0 for uninjured; -100 for maximally impaired gait).
2.5 Tissue preparation and RNA extraction
Nerve lesions were produced on the right side and the intact contralateral nerve served as the uninjured control. At the appropriate time points, nerves were harvested and processed for histology or immediately frozen in liquid nitrogen for subsequent extraction of RNA.
2.6 Primary SCs culture
SCs primary cultures were obtained from newborn SD rat sciatic nerves and the fibroblasts were removed with fibronectin-specific antibody and rabbit complement (Thermo Fisher Scientific, Waltham, MA, USA) according to the method previously described (28). Primary culture of SCs was maintained in DMEM containing 10% FBS at 37°C under humidified 5% CO2. SCs growth was observed under an inverted phase contrast microscope (Thermo Fisher Scientific). SCs cultures were passaged no more than 5 times before conducting experiments. SCs (1×105) and macrophages (1×105) were co-cultured in DMEM medium containing 10% FBS using transwell inserts (BD Biosciences, CA, USA).
2.7 Isolation of SCs-derived exosomes (SCs-Exo) and identification
The exosomes were isolated from SCs using Exoquick Reagent (SBI) as the manufacturer’s instruction. Briefly, conditioned media were incubated with Exoquick reagent (5:1) for over 12 h, centrifuged at 1,500 g for 30 min and pelleted exosomes were resuspended in 100 mL PBS.
Transmission electron microscopy (TEM) was used for morphological observation. The exosome samples were prepared as described above. For TEM, briefly, the exosomes were fixed with 2.5% glutaraldehyde overnight at 4°C. The solution was centrifuged at 100,000 × g to remove the glutaraldehyde, and the exosomes were washed three times with PBS. Then, the exosomes were stained with 3% phosphotungstic acid aqueous solution and fixed on copper mesh formvar grids. A transmission electron microscope (JEM-1010; JEOL, Tokyo, Japan) was used to detect the exosomes. Moreover, to analyze the distribution of particle size of the exosomes, a partial sample of exosomes was added to the sample cell without dilution. All operations were conducted in accordance with the instruction manual of the ZetaView® NTA technique (Particle Metrix, Germany). 10 µg of exosomes (re-suspended in PBS) were used to treat macrophages according to previous reports (2) and our preliminary results.
2.8 Exosome labeling and tracking
Exosomes were isolated from the culture medium and labeled with PKH67 Green Fluorescent membrane linker dye (Sigma-Aldrich, St. Louis, MO, USA) according to manufacturer's instructions. Then, the labeled exosome pellets were resuspended and added to the unstained macrophages for exosomes uptake studies. After incubation for 30 min, 2 h, or 12 h at 37 ℃, cells were observed by fluorescence microscopy.
2.9 Immunofluorescence
Rat sciatic nerves were fixed in situ with 4% PFA for 10 min, dissected, embedded in O.C.T. Compound (Tissue Freezing Medium; Solarbio, Shanghai, China), and frozen at -80 ℃. Sciatic nerve cryosections (5-µm thick) were first incubated with acetone for 10 min at -20 ℃, washed in PBS/0.1% Tween 20, blocked for 30 min at room temperature (RT) in blocking buffer (0.3% Triton X-100/10% Goat serum/phosphate buffer saline ¼ PBS), and incubated with primary antibodies overnight at 4 ℃ in blocking buffer. Cryosections were first incubated with 70% Ethanol for 5 min at RT, washed with PBS and incubated for 40 s with 40 mg/ml Proteinase K, before incubation with blocking buffer. Sections were then washed 3 times in blocking buffer and secondary antibodies were incubated for 1 h at RT in the dark. Sections were washed again, incubated with DAPI for 5 min at RT, washed and mounted in Citifluor (Agar Scientific).
Primary antibodies used for Immunofluorescence were as follow: Neurofilament (1:1000, Abcam, ab8135), SCG10 (1:500, Abcam, ab115513), IBA1 (1:100, Abcam, ab178847), NF200 (1:100, Abcam, ab82259), CD68 (1:100, Abcam, ab125212). All secondary antibodies were also purchased from Abcam. Photos were acquired using a Leica TCS SP-II confocal microscope.
2.10 (Fluorescence in situ hybridization) FISH
The subcellular localization of lncARAT was assessed using FISH assay with RiboTM lncRNA FISH Probe Mix (Green) (RiboBio, Guangzhou, China). The sciatic nerve tissue sections were fixed with 4% Paraformaldehyde (PFA). Slides were pretreated with protease K (2 µg/mL), glycine and acetic anhydride, followed by pre-hybridization for 1 h and hybridization at 42°C with probes (250 µL, 300 ng/mL) against lncARAT. Finally, slides were stained with phosphate buffered saline with DAPI (Sigma-Aldrich). Finally, 5 random fields acquired from each slide were observed and photographed by a fluorescence microscope.
2.11 Semithin and ultrathin sections and electron microscopy
After killed with 30mg/kg 3% pentobarbital sodium i.p., the rat’ sciatic nerves were fixed in situ with 3% paraformaldehyde and 0.15% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4. Fixed tissues were post-fixed in 2% osmium tetroxide, dehydrated through a graded acetone series as described previously, and embedded in Spurr’s resin (Electron Microscopy Sciences, EMS). Semithin sections were stained with 1% Toluidine blue for analysis at the light microscope, and ultrathin sections (70-nm thick) were made. All analyses were done at 5 mm distal to the lesion site. No contrasting reagent was applied. Images were acquired using a Philips CM 100 BIOTWIN equipped with a Morada sidemounted digital camera (Olympus).
2.12 RNA pull-down and RNA immunoprecipitation (RIP)
RNA pull-down assays were performed with the Magnetic RNA-Protein Pull down Kit (Thermo Scientific) according to the manufacturer’s instructions. 3 µg biotin-labeled RNA and 1 mg of extract were used in each pull down assay. The retrieved protein was separated on polyacrylamide gel electrophoresis (PAGE) gels and visualized by standard immunoblotting.
RIP assay was performed using the EZ-Magna RIP kit (Millipore, MA, USA). In brief, 1×107 cells were harvested and lysed with RIP lysis buffer with one freeze-thaw cycle. Cell extracts were co-immunoprecipitated using anti-KMT2A (ab272023), anti-KMT2B (ab104444), anti-KMT2D (ab224156) or Ago2 (ab226943) antibody, and the retrieved RNA was subjected to qPCR analysis.
2.13 ChIP and ChIRP analysis
The ChIP experiments were performed using the ChIP kit (Millipore, MA, USA). A total of 1×106 cells were fixed in 1% formaldehyde at room temperature for 10 min, and the nuclei were isolated with nuclear lysis buffer supplemented with a protease inhibitor. The chromatin DNA was sonicated and sheared to lengths between 100 and 200 bp. The sheared chromatin was immunoprecipitated at 4°C overnight using an anti-lncARAT antibody, anti-KMT2A antibody or anti-H3K4me3 antibody (Abcam, MA, USA). Normal mouse IgG was used as the NC. The ChIP-qPCR primers are listed in Supporting Table S9.
The Magna ChIRP RNA Interactome Kit was purchased from Millipore (Millipore, MA, USA) and used according to the manufacturer’s instructions. In brief, the probes were designed using a single-molecule FISH online designer, were biotin-labeled at the 3′end and were divided into an “odds” or an “even” groups. A total of 2×107 cells were cross-linked for each hybridization reaction. Then, the cell lysate was sonicated to shear the chromatin into 100–200 bp fragments. The sonicated cell lysates were hybridized with a mixture of biotinylated DNA probes for 4 h at 37°C. Then, the binding complexes were recovered using streptavidin-conjugated magnetic beads. Finally, DNA, RNA and protein were eluted and purified from the beads. The probes used in the ChIRP assay are listed in Supporting Table S9.
2.14 Lentivirus construction and RNA interference (RNAi)
The Lentivirus harboring lncARAT (Lv-lncARAT) or SOCS2 (Lv-SOCS2) cDNA was produced by GenePharma (Shanghai, China). An unrelated shRNA without any match with the rat genomic sequence was used as a control (Lv-Cont). Small interference RNA (siRNAs) for specifically inhibiting lncARAT (si-lncARATs), SOCS2 (si-SOCS2s), or short hairpin RNA (shRNAs) for specifically inhibiting lncARAT (sh-lncARATs) was designed and produced by GenePharma (Shanghai, China), and transfected with HiPerFect Transfection Reagent (Qiagen, CA, USA). The siRNA and shRNA sequences were showed in Supporting Table S9.
2.15 Reverse transcription-quantitative PCR (qPCR)
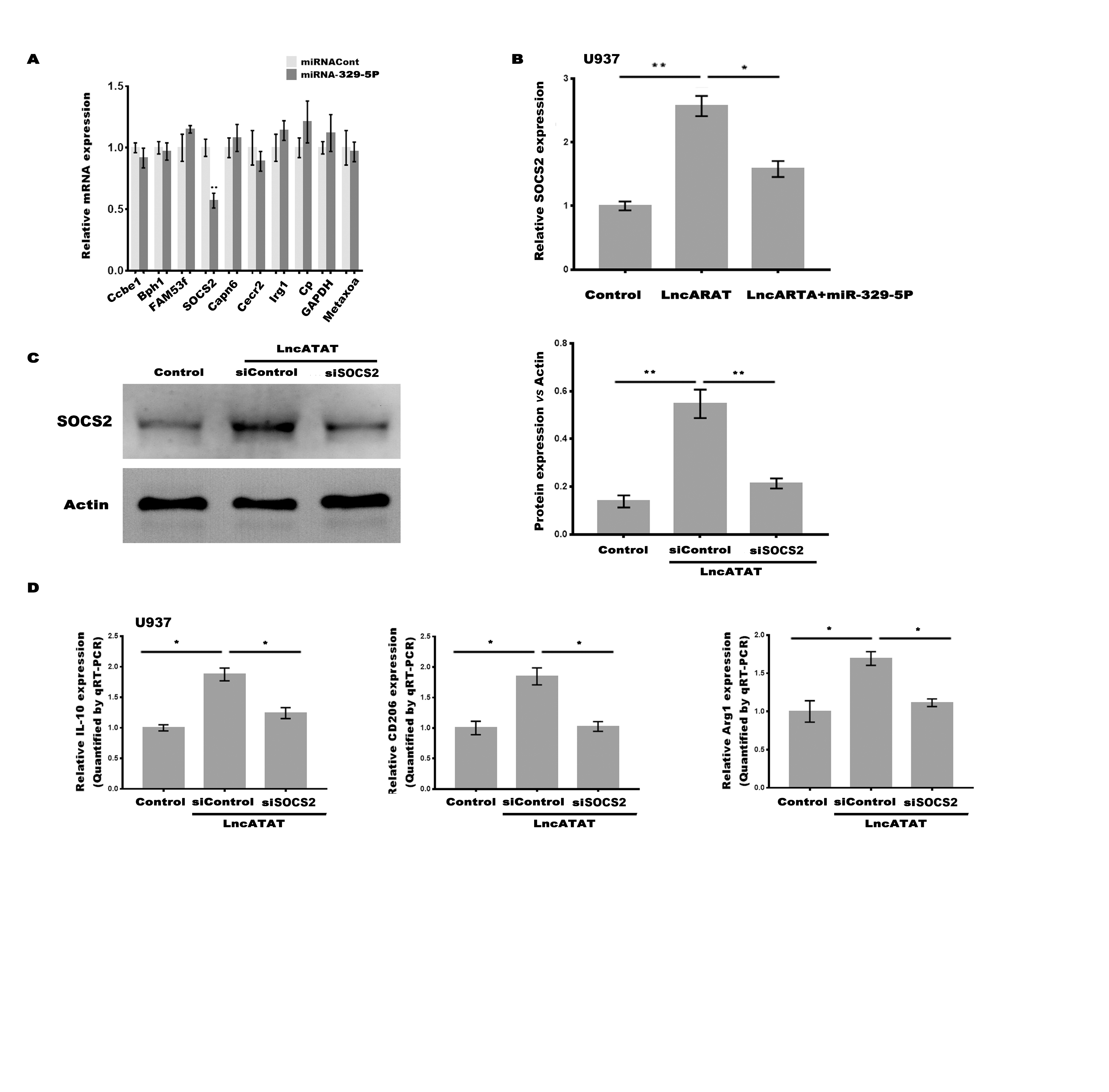
Total RNA from tissues and cells was extracted using a TRIzol® kit (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instruction. Reverse transcription was carried out using a PrimeScript RT reagent Kit (Takara Bio, Inc., Tokyo, Japan). The RT system of 10 µL were carried out according to the manufacturer’s instruction. RT conditions were 37 oC for 15 min and 85 oC for 5s. mRNA, lncRNA and miRNA expression levels were determined using the SYBR Green Supermix (Invitrogen) on the Applied Biosystems 7300 real-time PCR system. Thermocycling conditions were 95 oC for 10 min, following by 35 cycles of 95 oC for 10s, 58 oC for 15s and 72 oC for 20s, and final 72 oC for 20 min. β-actin was used as the internal control. All qPCR experiments were performed at ≥ 3 times, and the primer sequences were shown in Supporting Table S9.
2.16 Western blot analysis
Total protein was extracted from nerves or macrophages using RIPA buffer (Solarbio). The concentrations of the extracted nuclear and cytoplasmic fractions were quantified using a BCA protein assay kit (Pierce). A total of 50 µg protein per sample was separated by SDS-PAGE (10%) and then transferred to a PVDF membrane, prior to blocking with 5% non-fat milk in 1xTBST, overnight at 4°C. The membrane was then incubated with anti-CCL2 (1:2000; ab25124; Abcam, Cambridge, MA, USA), anti-iNOS (1:500; ab15323; Abcam), anti-Arg1 (1:1000; ab91279; Abcam), anti-CD206 (1:1000; ab125028; Abcam), anti-GAPDH (1:10000; ab181602; Abcam), anti-SOCS2 (1:1000; PA5-17219; Thermo Fisher Scientific) and Actin (1:5000; ab179467; Abcam) primary antibodies overnight at 4 oC. After washing 3 times in 1xTBST, the membranes were incubated with the corresponding HRP-conjugated secondary antibody (1:5000; ab205718; Abcam) for 1 h at room temperature. The immunoreactive proteins were visualized by an enhanced chemiluminescence reaction, and the band density was calculated by ChemiDoc™ XRS + Imaging system (Bio-Rad).
2.17 Dual-luciferase reporter assay
Recombinant plasmids of pGL3-lncARAT-Wt, pGL3-lncARAT-Mut, pGL3-SOCS2-3’UTR-Wt, and pGL3-SOCS2-3’UTR-Mut were constructed (Supporting Table S9). 0.5×105 HEK293 cells were plated into 48-well plate and co-transfected with 50 nM of miRNA-329-5p (or miRNA control), 20 ng of either pGL3-lncARAT-Wt, pGL3-lncARAT-Mut, pGL3-SOCS2-3’UTR-Wt, or pGL3-SOCS2-3’UTR-Mut, and 2 ng of pRL-TK (Promega, Madison, WI) using HiPerFect Transfection Reagent (Qiagen). pRL-TK was served as the internal control. HEK293 cells were collected and lysed 48 h after transfection and the luciferase activity was assessed using the Dual-Luciferase Reporter Assay System (Promega).
2.18 Cell migration
Cell migration was assessed using transwell chamber (BD Biosciences, 8-µm pore size, 24-well). Briefly, macrophages (1×104) suspended in 200 mL serum-free medium were seeded into the upper chamber, and 1×104 SCs in 800 mL medium containing 10% FBS were added to the bottom chamber. After 24 hours of culture, cells were then stained with 0.1% crystal violet for 30 minutes, and nonmigrating cells were removed. Six visual fields were randomly chosen to calculate the number of migrated cells.
2.19 Macrophage depletion
Macrophages were depleted through i.p. administration of clodronate liposome in rats according to previously reported method (29). In brief, 0.8 ml clodronate liposome (7 mg/ml) (Clodrosome, Encapsula NanoSciences, USA) was injected into rats with CSN. Control rats received i.p. administration of equal volume of PBS liposome.
2.20 Statistical analysis
All data were presented as the mean ± standard deviation (SD). The significance of differences between groups were assessed by Student’s t test and multiple group comparisons were performed using one-way ANOVA followed by the Scheffé test. SPSS 20.0 statistical software was applied for statistical analyses. p < 0.05 was statistically significant.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}