Patients and specimens
24 paired breast cancer tissues and adjacent normal tissues were collected from The First Medical Center of PLA General Hospital. All participants provided their written informed consent. Tissue samples were frozen in liquid nitrogen, storing at − 80℃ till RNAs were extracted.
Cell lines and culture conditions
All Human breast cancer cell lines, obtained from the Institute of Biochemistry and Cell Biology of the Chinese Academy of Sciences (Shanghai, China), were cultured in RPMI Medium 1640 (GIBCO, Grand Island, USA) with 1% penicillin/streptomycin and 10% FBS. The cells were incubated at 37℃ with 5% CO2 in a humidified incubator.
Fluorescence in situ hybridization (FISH)
MDA-MB-231 and MCF-7 cells were fixed with 4% paraformaldehyde at room temperature for 15 min, and 0.5% TritonX-100 was used for permeabilization at 4 °C for 20 minutes. Then, cells were incubated with the FISH probe of PCDHB17P at 55℃overnight. After hybridization, the slides were subjected to washing and dehydration. DAPI was performed to counterstain nuclei. The cells were captured using a fluorescence microscope (Olympus, Tokyo, Japan) and were merged using Adobe Photoshop 6.0 software.
Cell transfection
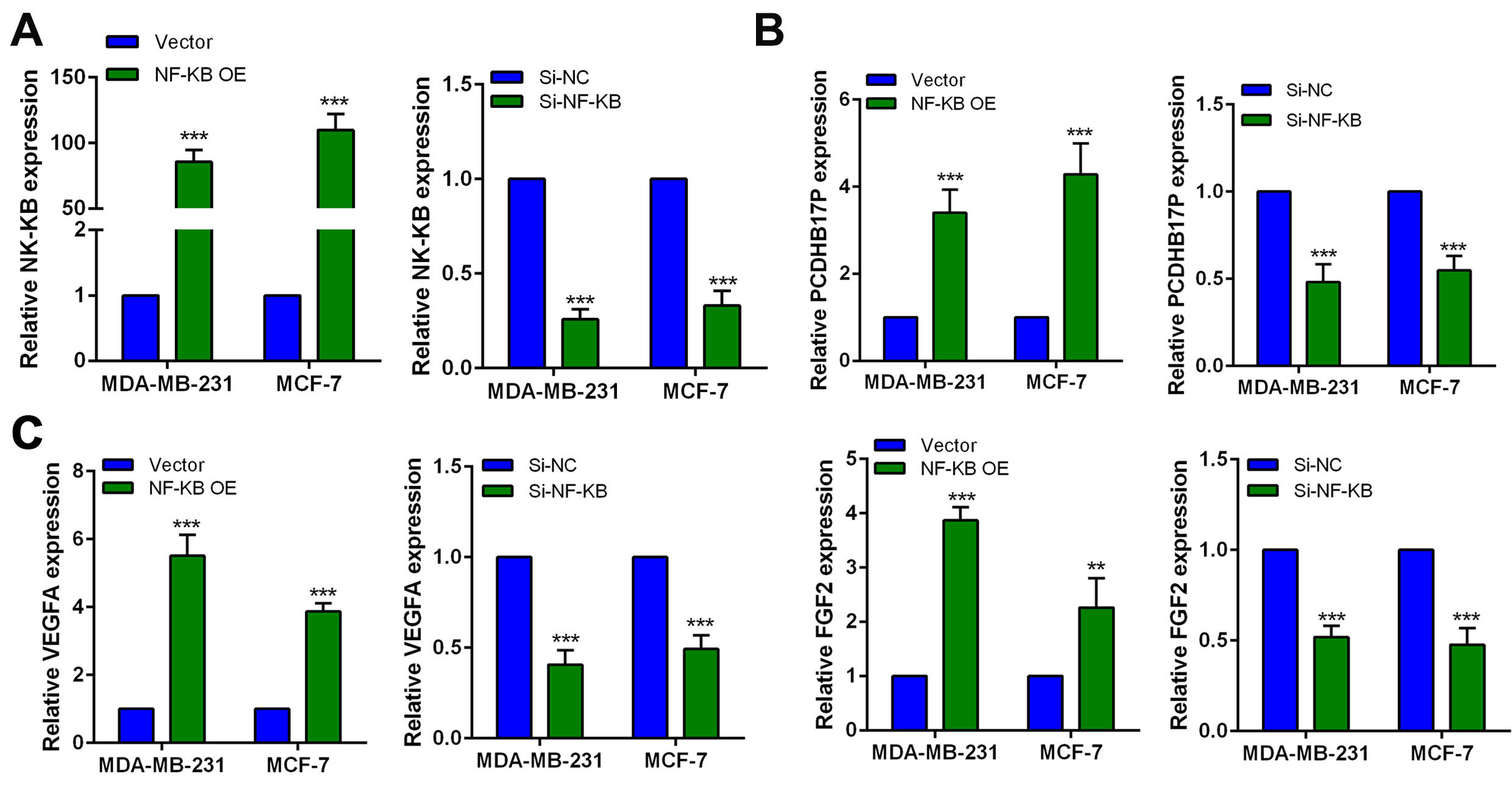
According to the manufacturers’ protocol, MDA-MB-231 and MCF-7 cells were severally transfected with each of the following by utilizing Lipotransfectamine 3000(Invitrogen, California, USA). PCDHB17P-specific short hairpin RNA (sh-PCDHB17P-1, sh-PCDHB17P-2) and negative control (sh-NC) were produced by RiboBio (Guangzhou, China), along with pcDNA3.1 vector containing PCDHB17P and empty vectors, were all from GenePharma (GenePharma, Shanghai, China). MiR-145-3p mimics, miR-145-3p inhibitors and their corresponding miR-NCs were synthesized by RiboBio (Guangzhou, China).
Quantitative real-time PCR analysis (RT-qPCR)
Total RNA was extracted from tissue specimens or cells using TRIzol reagent (Invitrogen, California, USA). The nuclear RNA and cytoplasmic RNA were separated by Purification Kit (Norgen, Thorold, Canada). Complementary DNA (cDNA) was synthesized with PrimeScript RT Reagent Kit (TaKaRa, Tokyo, Japan). The RT-PCR was accomplished using SYBR Green real-time PCR kit (TaKaRa, Tokyo, Japan). GAPDH and U6 expression were respectively utilized as the endogenous control for mRNA/lncRNA and miRNA. Gene expression levels were quantified through 2 −ΔΔCt method. The primer sequences used in RT-qPCR are listed in addifile file1: Table S1.
HUVEC tube formation assay
BD Matrigel™ Matrix was diluted with the serum-free DMEM medium at a ratio of 1:1. Then, 200 µL of mixture was added onto 24-well plates and polymerized for 1 h at 37℃. HUVEC suspension was added into the solidified gel. After 6 h, tube formation was observed and captured with microscopy.
Wound Healing Assay
The cells were seeded in 6-well plates and incubated at 37℃ until the cells reached 90% confluence. Briefly, cell layers were scratched by 200 µL sterile pipette tip and cells were then maintained in DMEM at 37℃and 5% CO2. Cell migration was observed and calculated at 0 h and 24 h with an invert microscope.
Western blotting analysis
The cells was collected and dissolved in RIPA lysis buffer (Beyotime, Jiangsu, China) at 4℃, the extracted proteins were separated by SDS-polyacrylamide gel (10%). After electrophoresis, proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Millipore, Massachusetts, USA). Then, PVDF membranes were blocked with 5% skimmed milk for 2 h with gentle shaking, and incubated overnight at 4 °C with the following primary detection antibodies: Anti-MELK (1:2000, Abcam, Cambridge, England), Anti-p65 ( 1:2000, CST, Boston, USA), Phospho-Akt ( 1:2000, CST, Boston, USA), Anti-p-P65 (1:1000, Abcam, Cambridge, England), anti-p-IKKβ(1:2000, CST, Boston, USA) and anti-p-IKKβ(1:2000, CST, Boston, USA). The following day, the membrane was washed and incubated with horseradish peroxidase-conjugated secondary antibody (goat anti-rabbit, 1:5000; Wanleibio, Shenyang, Liaoning, China) for 1 h. and the proteins were detected using BeyoECLPlus (Beyotime, Jiangsu, China).With GAPDH used as an internal reference, the relative expression of individual protein was calculated by the ratio of the gray value of the target protein to the internal reference band.
RNA immunoprecipitation (RIP)
The EZMagna RIP Kit (Millipore, Bedford, USA) was used for RIP according to the manufacturer's protocol. The cells were lysed and incubated in an ice bath with protein A magnetic beads, and centrifuged to collect the supernatant. The cell extract was co-precipitated by incubation with the antibody. The magnetic beads were washed and re-suspended in RIP Wash Buffer. Then, added appropriate antibodies to each group. The magnetic bead-antibody complex and the cell extract were incubated overnight at 4℃. After that, the magnetic bead-protein complex was harvested. The sample was digested with proteinase K to extract RNA for subsequent PCR detection.
Transwell assay
Transfected cells were collected with serum-free medium. Approximately 20,000 cells were added into the upper chamber of a transwell (Corning, New York, USA) with or without Matrigel (BD, New Jersey, USA) to evaluate invasion or migration functions. Medium with 15% FBS was placed to the lower chamber. 48 h later, cells that had invaded were fixed by 4% paraformaldehyde and dyed utilizing crystal violet (Beyotime, Jiangsu, China). Cells were observed and captured with microscopy.
Luciferase reporter assay
The sequences of PCDHB17P or MELK 3'-UTR containing the putative binding sites of miR-145-3p were subcloned into a pGL3 Dual-luciferase vector (Promega, Madison, USA). The luciferase reporter plasmids were co-transfected into MDA-MB-231 and MCF-7 cells with miR-145-3p mimics or MELK OE and the negative control. 48 h after transfection, luciferase signals were measured with the Dual Luciferase Reporter Assay System (Promega, Madison, USA) according to the manufacturer’s instructions.
Chromatin immunoprecipitation (ChIP)
The ChIP Assay Kit (Millipore, Bedford, USA) was used for ChIP according to the manufacturer's protocol. Briefly, the cells were collected and sonicated to generate chromatin samples with average fragment sizes of 100–500 bp. Then, immunoprecipitated with anti-MELK (Abcam, Cambridge, England) or anti-IgG (Abcam, Cambridge, England) antibodies at 4℃ overnight. The immunoprecipitated DNA was eluted and purified for the subsequent qPCR analysis.
Immunohistochemistry (IHC)
The tissue samples were placed into citrate buffer and heated in a microwave oven. Slides were then incubated with anti-VEGFA (1:100, Abcam, Cambridge, England), anti-MELK (1:500, Abcam, Cambridge, England) or anti-CD33(1:100, Abcam, Cambridge, England) at room temperature for 1 h. Following washing, each section was briefly counterstained using a catalyzed sign amplification system kit (Dako code k5007). The images were captured under the same conditions.
In vivo nude mouse models
Female BALB/c nude mice (aged 5 weeks) was bought from Guangdong Medical Laboratory Animal Center, and kept under specific pathogen-free conditions. 5 × 106 transfected breast cells were injected subcutaneously into the flank of nude mice, 6 weeks later, the tumors were isolated,and subjected to Immunohistochemistry. For the lung metastasis model, 1 × 10 6 cells were injected into the tail veins of the mice. After 4 weeks, the mice were killed and the lung tissue were fixed and paraffin-embedded for hematoxylin and eosin (H&E) staining.
Statistical analysis
Data were analyzed by GraphPad Prism 6.0 and shown as mean ± standard deviations, unless otherwise specified. Differences between groups were analyzed by unpaired Student’s t-test between two groups and one-way ANOVA followed by Tukey comparison test in more than three groups. Statistical significance was defined as *p ≤ 0.05, **p < 0.005 and ***p < 0.001, respectively.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}