Human pluripotent stem cell (hPSC) culture
Human pluripotent stem cell (hPSCs) lines, Royan H5 (RH5; RRID:CVCL_9386), Royan H6 (RH6; RRID:CVCL_9387) [38] and CAG-human induced pluripotent stem cell (CAG-hiPSC) were obtained from the Royan stem cell bank, Tehran, Iran. To examine different cell seeding densities in co-culture, the huES4 PDX1-GFP cell was used [39] and expanded in DEF-CS 500 (Takara) [12], and passaged with TrypLE. Experiments with this line were conducted at DanStem and approved by De Videnskabsetiske Komiteer, Region Hovedstadenunder number H-4-2013-057. Cells obtained from the Royan stem cell bank were maintained on Matrigel (Sigma-Aldrich)-coated dishes and hPSCs’ culture medium that consisted of Dulbecco’s modified Eagle’s medium/F12 (DMEM/F12) supplemented with 20% knockout serum replacement (KoSR), 0.1 mM non-essential amino acids (NEAA), 1% penicillin/streptomycin, 1% glutaMAX, 0.1 mM β-mercaptoethanol (all from Invitrogen), 1% insulin-transferrin-selenium (ITS, Life Technologies), and 100 ng/ml basic fibroblast growth factor (bFGF, Royan Biotech, Iran). The medium was changed every day and cells were passaged every 6-8 days by collagenase/dispase (1:2) solution [collagenase IV (0.5 mg/ml, Invitrogen): dispase (1 mg/ml, Invitrogen)] for maintenance. To induce differentiation, hPSCs were singled by trypsin-EDTA (Invitrogen), treated with 10 µM Y27632 (Sigma-Aldrich) as a ROCK inhibitor (ROCKi), and seeded at a density of 200 × 103 cells/cm2. The differentiation process was started when cells reached 80-90% of confluence (i.e. after 24-48 h).
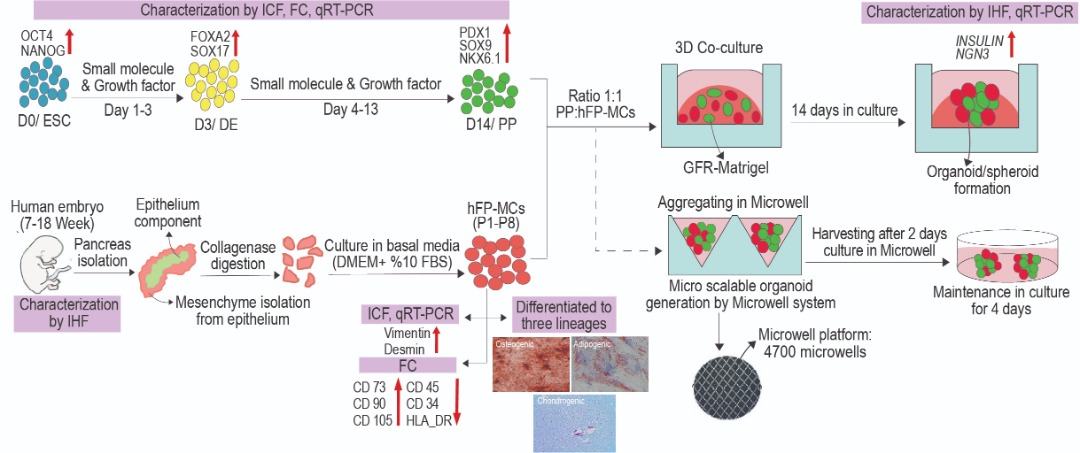
Generation of pancreatic progenitor cells (PPs) from hPSCs
Pancreatic progenitor cells (PPs) were differentiated from hPSCs within 14 days following a standard protocol [40] with some modifications. The designed stepwise protocol included definitive endoderm (DE), posterior foregut (PF), early PP, and PP stages, respectively. For endoderm differentiation, the hPSCs were cultured for three days in endoderm base medium containing RPMI-1640 (Invitrogen), 1% glutaMAX, 1% penicillin/streptomycin, 1% NEAA, 1% bovine serum albumin (BSA; Sigma-Aldrich), and 0.1 mM β-mercaptoethanol. In order to achieve mesendoderm on the first day of differentiation, 100 ng/ml Activin A (Sigma-Aldrich) and 2 μM Chir99021 (Stemgent) were added to the medium. To induce DE formation on days 2 and 3 of differentiation, Chir99021 was removed, and cells were induced by 100 ng/ml Activin A, 50 μg/ml ascorbic acid (AA, Sigma-Aldrich), and 5 ng/ml bFGF. Next, to achieve PPs, the differentiated endoderm cells were exposed to DMEM/F12 supplemented with 1% B27 minus vitamin A (Gibco), 1% glutaMAX, 1% penicillin/streptomycin, 1% NEAA, and 1% BSA, for 11 additional days. The base medium was supplemented with 50 ng/ml fibroblast growth factor 10 (FGF10; Royan Biotech) for 2 days, and afterward, 0.25 μM KAAD-cyclopamine (CYC, TRC), 100 nM PDBu (Tocris Biosciences), 2 μM retinoic acid (RA, Sigma-Aldrich), 200 nM LDN193189 (Stemgent), 50 ng/ml FGF10, and 50 μg/ml AA were added to the base medium on days 6 and 7 to achieve early PPs. Finally, from day 8 onwards, 50 ng/ml EGF (Sigma-Aldrich), 200 nM LDN193189, 10 mM nicotinamide, 100 nM RA, 50 ng/ml FGF10, and 50 μg/ml AA were added to the base medium. At the end of days 3 and 14 of differentiation, cells were harvested and analyzed by quantitative real time-PCR (qRT-PCR), immunostaining, and flow cytometry. To examine different cell seeding densities in co-culture, the PPs were differentiated from huES4 PDX1-GFP cell line based on a modified published protocol [41].
Human fetal donation, pancreas retrieval, and mesenchyme isolation
Human fetal pancreatic-derived mesenchymal cells (hFP-MCs) were obtained from Endocrinology and Metabolism Research Institute, Tehran University of Medical Sciences, Tehran, Iran [42]. The human fetal pancreases were harvested from legally aborted fetuses at weeks 7-9 and 14-18 of gestation following obtaining maternal written informed consent. The ethics committee approved the use of fetal tissues for research (No. EC-00264) of the Endocrinology and Metabolism Research Institute, Tehran University of Medical Sciences, Tehran, Iran. Briefly, the donated fetuses were immersed in 5% povidone-iodine solution, followed by washes in phosphate buffered saline (PBS). Afterward, the fetal pancreas was harvested by a midline laparotomy, pulled out, and dissected from surrounding tissues. Then, the fetal pancreas was washed several times and minced into small pieces. Ultimately, short enzymatic digestion was performed using 1 mg/ml collagenase for 15 min. Digestion was stopped by cold PBS, and the vials were placed for 10 min on ice. After discarding the supernatant, the pellet containing pancreatic epithelial and mesenchymal cells was re-suspended in culture media containing Dulbecco’s modified Eagle’s medium/low glucose (DMEM/low glucose, Thermo Scientific, USA) and 15% fetal bovine serum (FBS, Invitrogen), and kept in a 25cm2 flask. After 2 days, unattached tissue fragments were discarded, and fresh media was added to attach MCs. For the second pancreatic mesenchymal subculture, media was changed to DMEM-LG and 10% FBS.
For examining different cell seeding densities in co-culture, experiments were performed at DanStem with human fetal pancreases obtained from material available following elective termination of pregnancy between weeks 8 and 10 at the Departments of Gynaecology at Copenhagen University Hospital (Rigshospitalet) and Hvidovre Hospital, Denmark. The regional ethics committee approved this study (permit number H-1-2012-007), and women gave their informed written and oral consent. None of the terminations were for reasons of the pathology of pregnancy or fetal abnormality. Fetal age was determined by scanning crown-rump length and by evaluation of foot length [43].
The bone marrow mesenchymal stem cells (BM-MSCs) were obtained from the Royan stem cell bank.
Expansion and characterization of hFP-MCs and BM-MSCs
hFP-MCs from three 7-9-week post-conception (WPC) (named as MC1-3) and three 14-18 WPC (named as MC4-6) fetal samples were cultured in standard MCs culture medium that contained DMEM-LG supplemented with 10% FBS, 2 mM glutaMAX, 0.1 mM NEAAs and 1% penicillin/streptomycin. The medium was changed every two days [42]. When MCs confluence became 70-80%, cells were sub-cultured at a ratio of 1:3 by trypsin-EDTA. The hFP-MCs were passaged up to passage number 9. Based on the international society for cellular therapy’s (ISCT) minimal criteria, MCs from MC4-6 subcultures 4-6 were examined for mesoderm-lineage differentiation capacity and different CD marker expression. The expression of DESMIN and VIMENTIN genes (as mesenchyme markers) from different passages of MC1-6 was also evaluated by qRT-PCR.
hESC-PPs co-culture with hFP-MCs in a 3D culture system
To evaluate hFP-MCs effect on PPs, day-14 hESC-PPs were co-cultured with hFP-MCs or BM-MSCs (control group) on GFR-matrigel-coated 96-well plate (Corning). Here, three experimental groups were considered: 1) hESC-PPs cultured solely in Matrigel (hESC-PP), 2) hESC-PPs co-cultured with hFP-MCs (CO PP/hFP-MC), and 3) hESC-PPs co-cultivated with BM-MSCs (CO PP/BM-MSC). In this study, a 3D culture system (sandwich method) was used for co-culture where different cell densities of hESC-PPs: hFP-MCs or hESC-PPs: BM-MSCs at a ratio of 1:1, were examined. The pancreatic endoderm (PE) medium used to maintain PPs in their progenitor state contained DMEM/F12, 1% penicillin-streptomycin, 1% B27 culture supplement (Gibco), 64 ng/ml bFGF, and 10 µM Y-27632 (only on the first day) [44]. Initially, a mixture of 20 μl GFR-matrigel and PE medium (3:1 ratio) was added to each well and incubated for 15 min to solidify before cell seeding. hESC-PPs and hFP-MCs or BM-MSCs were mixed in GFR-Matrigel diluted 3:1 with PE medium and transferred onto the pre-existing solidified GFR-matrigel coated plate. Afterward, the plate was centrifuged, and 100 μl of PE medium was added to each well and kept for 14 days in culture. The medium was renewed every 3-4 days.
Agarose microwell chip preparation
Non-adherent agarose microwell chips were fabricated by mold-replication technology as previously described [45]. Briefly, AggreWell 400 plate (Stem Cell Technologies, France) was used as a template. Each well contained structured polydimethylsiloxane (PDMS) surface with a standard array of 4700 pyramidal microwells with a diameter of 400 µm. The PDMS mold was peeled off after curing of AggreWells. Then, 2 ml/well of 2% (W/V) ultrapure agarose (Invitrogen) solution was poured into standard 6-well plates, and sterilized PDMS mold was placed carefully on top of the liquid and allowed to swim up. When the mold was gently removed after agarose solidification, a mirror-inverted patterned agarose surface was observed.
Dual cell pancreatic aggregate (DC-PA) generation in 3D agarose microwell chip
For cell aggregate formation, cell suspension containing a total of 235 × 104 cells/well (at a seeding density of 500 cells/microwell) of PPs and hFP-MCs (at different ratios) was seeded on agarose microwell chips. Afterward, the plate was centrifuged for 4 min at 1200 rpm, in order to capture the cells in the microwells. Next, the plate was checked under a light microscope to confirm that the cells were distributed among the microwells; cells were then incubated in PE medium at 37°C with 5% CO2 and 95% humidity. After 48 h incubation, DC-PAs were aspirated from the microwells and placed in 30mm ultra-low attachment plates with a density of 1500 aggregates per 10 cm2 in the same maintenance medium used during aggregate formation. The images of DC-PAs formed were obtained daily using a digital camera to measure the diameter and number of the aggregates and were quantified by ImageJ. Aggregates size and number are reported as mean ± SEM.
Flow cytometry analysis
Flow cytometry was performed to quantify DE and PPs differentiation efficiency on days 3 and 14 of differentiation. Briefly, cells were dissociated by trypsin/EDTA for 5 min at 37˚C and washed with PBS. After centrifugation at 1500 rpm for 5 min, the cell pellet was fixed, permeabilized, and blocked. The cell pellet was re-suspended in primary antibody solution overnight, then treated with secondary antibody for 2 h at room temperature (RT). Quantification was performed by BD FACS Calibur flow cytometer (BD Biosciences), and the data were analyzed using Flowing Software (version 2.4.2). The primary and secondary antibodies used in this study are listed in Supplementary Table 1.
Immunostaining analysis
For immunocyto-staining, differentiated samples were fixed in 4% paraformaldehyde (PFA, Sigma‐Aldrich) at RT for 20 min. For immunohisto-staining, co-culture and DC-PA samples were fixed in 4% paraformaldehyde at RT overnight, processed, embedded in paraffin, and sectioned at a thickness of 5 μm using a microtome (MicromHM325, Thermo Scientific). After deparaffinization and rehydration of the sections, heat-mediated antigen retrieval was performed in citrate buffer (pH 6.0, Dako) or Target Retrieval Solution High (pH 9.0, Dako) in Retriever 2100 device according to the primary antibodies data sheet instructions. Samples and slides were washed in 0.05% Tween-PBS (Sigma-Aldrich), permeabilized in 0.2% Triton X‐100 (Sigma-Aldrich), blocked in 5% BSA (or 10% secondary antibody host serum) for 45-60 min and treated with primary antibodies at 4°C, overnight. Next, they were exposed to secondary antibodies at RT for 1 h in the dark. Cell nuclei were stained using 1 mg/ml 4’, 6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich). The samples were observed under Olympus BX51/IX71 fluorescence microscopes equipped with an Olympus DP72 digital camera or on microscopic plates of Zeiss LSM800 confocal microscope. For quantifying protein expression efficiency in the differentiation process of RH5-ESC and CAG-hiPSC cells, seven 40X frames were randomly captured from different areas of each sample in 3 independent repeats and counted by ImageJ software. The percentages of expression were calculated according to the number of positive cells per total cells stained with DAPI. The primary and secondary antibodies used in this study are listed in Supplementary Table 1. Three slides from each sample were stained with hematoxylin and eosin (H&E) for observing morphological structures.
RNA isolation and quantitative real time- PCR (qRT-PCR)
We performed qRT-PCR for evaluating proliferation, mesenchymal and pancreatic differentiation specific genes at the transcriptional level. First, total RNA was extracted by RNeasy Micro Kit (Qiagen) based on the manufacturer’s protocol. A spectrophotometer (WPA, Biowave II) was used to evaluate RNA concentration and purity. cDNA was synthesized from 1 μg of total RNA using Takara cDNA Synthesis Kit (Takara). Quantification of the transcript was performed by Applied Bio-system (ABI, Step One Plus) using SYBR Green reagent (Applied Biosystems) and 25 ng/μl of each sample. The relative expression levels of the target genes were normalized against GAPDH as the housekeeping gene and calculated based on 2^-∆∆Ct and 2^-∆Ct method for differentiation process samples and co-culture/ DC-PA samples, respectively. A “control” was used to diminish or eliminate the effect of the housekeeping gene expressed by the mesenchyme in the co-culture groups. The “control” contained a mix of hESC-PPs and hFP-MCs which each were separately kept in Matrigel along with the other groups for 14 days. At the end of the culture period, hESC-PPs and hFP-MCs were mixed together and considered as a single sample, “the hESC-PP”, for RNA extraction. The samples were collected from at least three independent biological replicates. The primer sequences are mentioned in Supplementary Table 2.
Live-dead assessment
To evaluate the viability of cells after co-culture and DC-PAs formation, live-dead assessment was performed using Fluorescein Diacetate (FDA, Sigma-Aldrich) and Propidium Iodide (PI, ThermoFisher) double staining. Here, 20 µl of FDA stock solution and 50 µl of PI stock solution were dissolved in 10 ml PBS [46]. For staining, FDA/PI solution was added directly to the culture, incubated for 5 min in the dark at RT and observed under Zeiss LSM800 confocal microscope.
hFP-MCs labeling with PKH26
For investigating the contribution and position of hFP-MCs in co-culture and in DC-PAs, hFP-MCs were labeled with the fluorescent vital dye PKH26 (Sigma-Aldrich) according to the manufacturer’s instructions. Briefly, 1 μl of PKH26 dye was diluted in 50 μl diluent C (from the manufacturer’s labeling kit) and added to 2-3×106 suspended hFP-MCs which were already diluted with 50 μl diluent C. Then, the suspension was incubated for 7-10 min at RT. The reaction was ended by adding 2 ml FBS, and cells were washed with DMEM/F12 to remove the excess dye. Finally, cells were observed under Zeiss LSM800 confocal microscope with an excitation wavelength of 551 nm and an emission wavelength of 567 nm.
Animals
ICR mice (Mus musculus) were housed at the University of Copenhagen with a standard 12 hours light/dark schedule. All experiments were performed according to ethical guidelines approved by the Danish Animal Experiments Inspectorate (Dyreforsøgstilsynet). For embryo staging, the day of vaginal plug was considered E0.5. All the embryonic stages used are stated in the main text or in the figure legends. Male and female embryos were used for all the experiments.
Statistical analysis
All data are presented as the mean ± SEM. Immunofluorescence images were quantified using ImageJ software. All statistical analyses of the data were done using Graphpad Prism 8 software by either unpaired t-test or one-way ANOVA followed by parametric and non-parametric tests. The differences among experimental groups were considered statistically significant when the p-value was less than 0.05.
{kind=link}