Low-dose DB increased cytotoxic effects of cisplatin on CR-GC cells
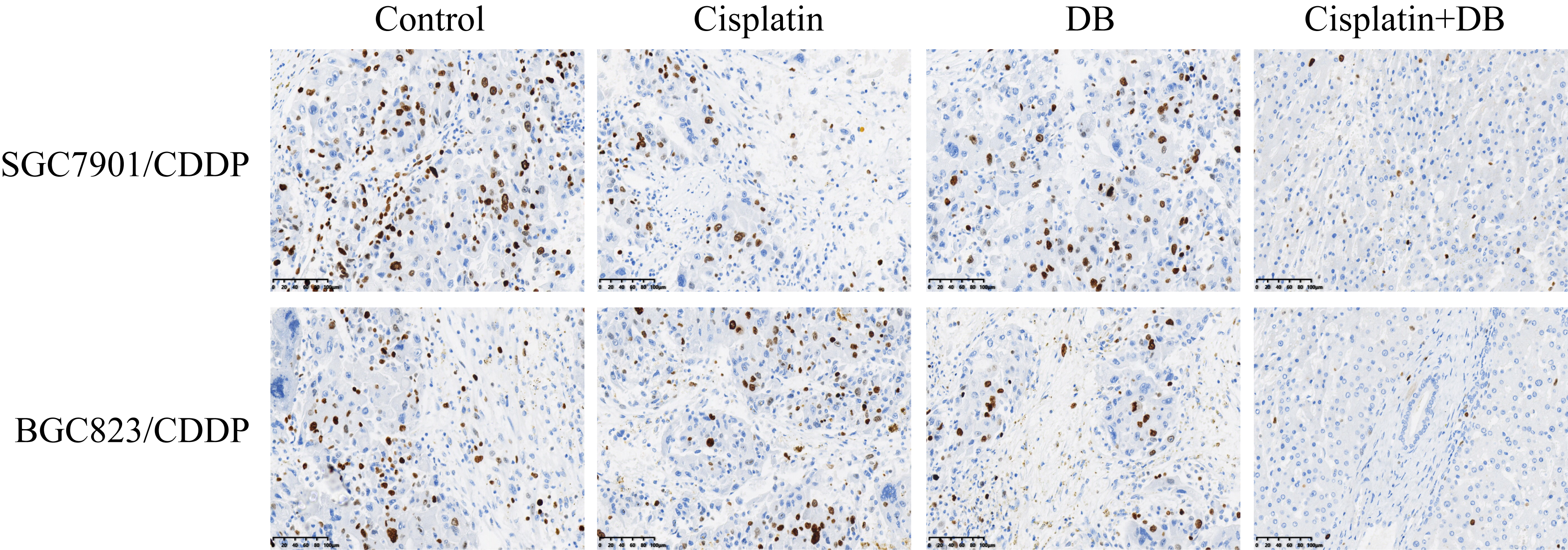
DB had been used for cancer treatment [34, 35], and the previous data from our team indicated that low-dose DB (12.5 µM) was advantageous for GC treatment to avoid DB-induced hepatotoxicity [34], which rendered the possibility that low-dose DB (12.5 µM) might be a novel agent to increase sensitivity of CR-GC cells to the traditional chemotherapeutic drugs, such as cisplatin. To achieve this, the established CR-GC cells (SGC7901/CDDP and BGC823/CDDP) were treated with cisplatin (20 µg/ml) combined with low-dose DB (12.5 µM) for 0 h, 24 h, 48 h and 72 h, respectively, according to our preliminary experiments. As shown in Fig. 1A-D, CR-GC cells were resistant to cisplatin stimulation, and low-dose DB alone did not influence cell proliferation and viability in CR-GC cells. Interestingly, DB combined cisplatin treatment significantly hindered CR-GC cell growth in vitro (P < 0.05, Fig. 1A-D). Consistently, by examining cell apoptosis, we found that low-dose DB triggered apoptotic cell death in cisplatin treated CR-GC cells (P < 0.05, Fig. 1E). Furthermore, the CR-GC cells (SGC7901/CDDP and BGC823/CDDP) were employed to establish xenograft tumor-bearing mice models, and we found that DB and cisplatin co-treatment significantly inhibited tumor weight (P < 0.05, Fig. 1F) and volume (P < 0.05, Fig. 1G, H) to hamper tumorigenesis of the CR-GC cells in vivo. In addition, the mice tumor tissues were collected and prepared, and our following results indicated that the expression levels of Cyclin D1 and CDK1 (P < 0.05, Fig. 1I, J), and Ki67 (Figure S1) were decreased, while Caspase-3 and Bax were increased (P < 0.05, Fig. 1K, L) by co-treating CR-GC cells with DB and cisplatin. The data in Fig. 1 and Figure S1 suggested that low-dose DB triggered apoptotic cell death to enhance the cytotoxic effects of cisplatin on CR-GC cells.
Induction of apoptosis and pyroptosis by low-dose DB in cisplatin-treated CR-GC cells
Since cisplatin inhibited cancer progression by inducing various types of cell death, including apoptosis, pyroptosis, ferroptosis and autophagy. To investigate by which types of cell death were induced by low-dose DB in cisplatin treated CR-GC cells, the CR-GC cells were pretreated with the inhibitors for pyroptosis (Necrosulfonamide, NSA) and apoptosis (Z-VAD-FMK), autophagy (Chloroquine) and ferroptosis (Ferrostatin-1, Fer-1), respectively. The results showed that merely NSA and Z-VAD-FMK, instead of Chloroquine and Fer-1, abrogated the inhibiting effects of DB-Cisplatin combined therapy on cell proliferation (P < 0.05, Fig. 2A, B) and viability (P < 0.05, Fig. 2C, D). Also, blockage of apoptosis and pyroptosis decreased cell apoptosis ratio in DB-cisplatin co-treated CR-GC cells (P < 0.05, Fig. 2E), suggesting that low-dose DB aggravated the inhibiting effects of cisplatin on CR-GC cells in a apoptosis- and pyroptosis-dependent manner. Further experiments were performed to uncover the detailed mechanisms, and we found that low-dose DB combined with cisplatin increased the expression levels of pyroptosis signatures (NLRP3, ASC, IL-1β and IL-18) (P < 0.05, Fig. 2F, G) and apoptosis (cleaved Caspase-3 and Bax) (P < 0.05, Fig. 2H, I) in CR-GC cells. Interestingly, the inhibitor for pyroptosis (NSA) decreased the expression levels of both pyroptosis and apoptosis signatures (P < 0.05, Fig. 2F-I), while the apoptosis inhibitor (Z-VAD-FMK) had little effects on cell pyroptosis in DB-cisplatin treated CR-GC cells (P < 0.05, Fig. 2F-I), indicating that low-dose DB induced cell apoptosis in cisplatin treated GC cells through triggering pyroptotic cell death. Next, the siRNA for NLRP3 were transfected into CR-GC cells to knock-down NLRP3 (P < 0.05, Fig. 2J, K), the results showed that NLRP3 ablation abrogated the promoting effects of low-dose DB-induced cell death in cisplatin treated CR-GC cells (P < 0.05, Fig. 2L), implying that low-dose DB sensitized CR-GC cells to cisplatin by activating NLRP3 mediated pyroptosis.
Enrichment of CSCs in CR-GC cells and continuous low-dose cisplatin stimulated CS-GC cells
Previous data indicated that CSCs were enriched in cancer cells after long-term cisplatin stimulation, which increased resistance of cancer cells to cisplatin by sustaining tumor heterogeneity. Expectedly, this study validated that CR-GC cells (SGC7901CDDP and BGC823CDDP) were prone to form spheres compared to their corresponding parental CS-GC cells (SGC7901 and BGC823) (P < 0.05, Fig. 3A), and the stem cell markers (SOX2, OCT4 and Nanog) were also upregulated in CR-GC cells instead of CS-GC cells (P < 0.05, Fig. 3B-E), suggesting that CSCs were enriched in CR-GC cells. Also, to validate the above results, according to the previous study, the parental CS-GC cells (SGC7901 and BGC823) were exposed to continuous low-dose cisplatin treatment to simulate the generation of acquired cisplatin resistant GC (ACR-GC) cells in vitro. As expected, we observed that continuous low-dose cisplatin pressure increased the spheroid formation abilities (P < 0.05, Fig. 3F), and promoted SOX2, OCT4 and Nanog expressions (P < 0.05, Fig. 3G-J) in ACR-GC cells, compared to the CS-GC cells. In general, analysis of the above data in Fig. 3 suggested that continuous cisplatin pressure induced CSCs properties in CR-GC and ACR-GC cells.
Low-dose DB regulated CSCs properties in CR-GC cells by inhibiting PD-L1
Since we had proved that low-dose DB sensitized CR-GC cells to cisplatin by triggering NLRP3 mediated pyroptosis, and cisplatin pressure induced generation of CSCs contributed to drug resistance of GC cells. Hence, we next investigated whether low-dose DB affected stemness of CR-GC cells. As shown in Fig. 4A, we found that low-dose DB inhibited spheroid formation abilities in CR-GC cells (P < 0.05). Also, the expression levels of SOX2, OCT4 and Nanog were decreased by low-dose DB in CR-GC cells (P < 0.05, Fig. 4B-E), suggesting that DB inhibited CSCs properties in CR-GC cells. Based on the information that PD-L1 regulated cancer cell stemness, we validated that PD-L1 was upregulated in CR-GC cells, compared to the corresponding CS-GC cells (P < 0.05, Fig. 4F). Interestingly, we proved that low-dose DB negatively regulated PD-L1 in CR-GC cells (P < 0.05, Fig. 4G), which enlightened us that low-dose DB might regulate cell stemness in CR-GC cells through PD-L1. To validate the above hypothesis, the PD-L1 overexpression vectors were successfully transfected into CR-GC cells (P < 0.05, Fig. 4H, I), and the results showed that upregulation of PD-L1 promoted the stemness signatures (SOX2, OCT4 and Nanog) expressions to improve CSCs properties in DB-treated CR-GC cells (P < 0.05, Fig. 4J-M), implying that low-dose DB inhibited CSCs enrichment in CR-GC cells by downregulating PD-L1.
Knock-down of PD-L1 triggered pyroptotic cell death in CR-GC cells through activating NLRP3 inflammasome
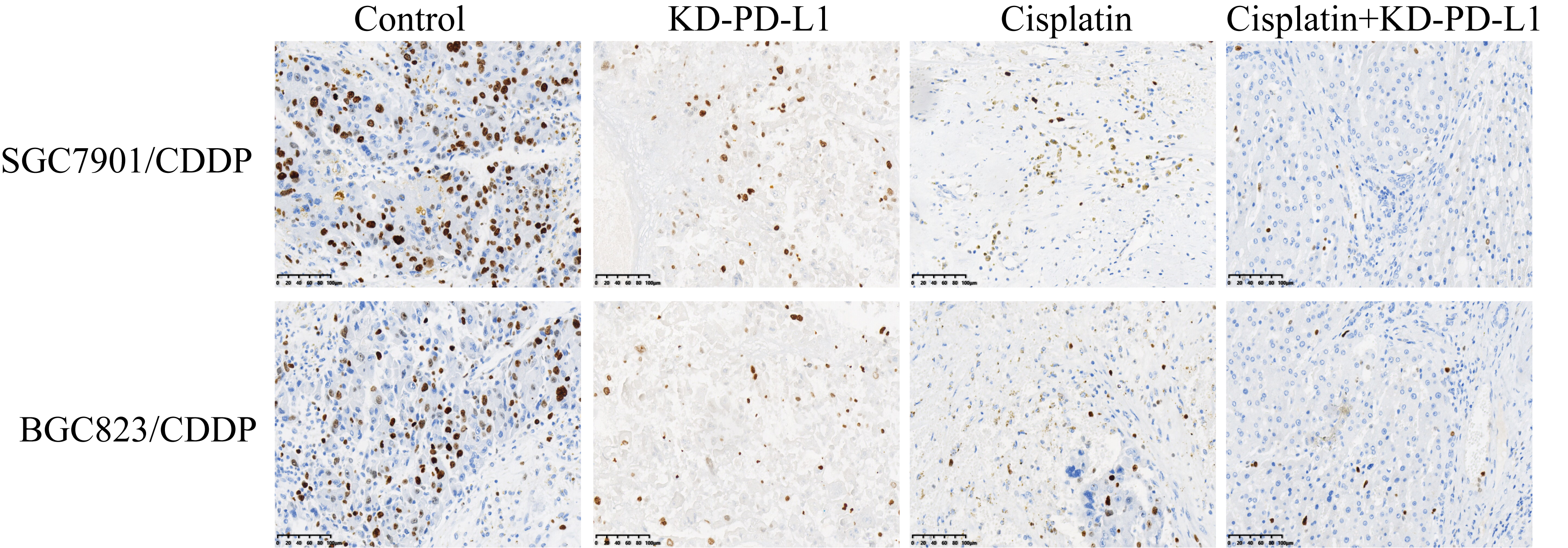
Aside from stemness, PD-L1 also directly regulated cell proliferation and death to modulate drug resistance in cancer cells, and recent data suggested that there existed a tumor-intrinsic PD-L1-NLRP3 signaling pathway in tumor cells [36], based on this, as shown in Fig. 5, our further experiments validated that silencing PD-L1 activated NLRP3 mediated pyroptosis in cisplatin treated CR-GC cells. Mechanistically, the PD-L1 overexpression and downregulation vectors were successfully delivered into CR-GC cells (P < 0.05, Fig. 5A, B), and we verified that PD-L1 negatively regulated NLRP3 in CR-GC cells (P < 0.05, Fig. 5A, B). In addition, knock-down of PD-L1 increased the expression levels of NLRP3, ASC, IL-1β and IL-18 to facilitate pyroptotic cell death in cisplatin treated CR-GC cells (P < 0.05, Fig. 5C, D), which were abrogated by silencing NLRP3 (P < 0.05, Fig. 5C, D), suggesting that targeting intrinsic PD-L1/NLRP3 pathway triggered cell pyroptosis in cisplatin-treated CR-GC cells. Also, either cisplatin or PD-L1 ablation alone promoted cell apoptosis in CR-GC cells to a very limit extent (P < 0.05, Fig. 5E), which were significantly enhanced by co-treating cells with cisplatin and PD-L1 downregulation (P < 0.05, Fig. 5E). Additionally, the promoting effects of cisplatin and silencing PD-L1 co-treatments on cell apoptosis in CR-GC cells were reversed by knocking down NLRP3 (P < 0.05, Fig. 5E). Furthermore, the xenograft tumor-bearing mice models were established, and we proved that knock-down of PD-L1 inhibited Cyclin D1 and CDK2 (P < 0.05, Fig. 5F, G), and Ki67 (Figure S2), while promoted Caspase-3 and Bax expressions (P < 0.05, Fig. 5H, I) to facilitate cell growth in CR-GC cells in vivo.
Low-dose DB enhanced the cytotoxic effects of cisplatin on CR-GC cells through downregulating PD-L1
Finally, as shown in Fig. 6, we found that low-dose DB sensitized CR-GC cells to cisplatin stimulation through targeting the intrinsic PD-L1/NLRP3 signaling pathway. Functionally, we found that cisplatin alone had little effects on PD-L1 and NLRP3 expressions (P > 0.05, Fig. 6A, B), while the promoting effects of cisplatin-DB co-treatment on NLRP3, ASC, IL-1β and IL-18 expressions were abrogated by overexpressing PD-L1 (P < 0.05, Fig. 6A-D), implying that DB triggered NLRP3-mediated pyroptotic cell death in cisplatin-treated CR-GC cells by downregulating PD-L1. Furthermore, induction of apoptotic cell death by cisplatin-DB co-treatment was reversed by overexpressing PD-L1 (P < 0.05, Fig. 6E), and the inhibiting effects of DB stimulation on cell proliferation (P < 0.05, Fig. 6F, G) and viability (P < 0.05, Fig. 6H, I) in cisplatin-treated CR-GC cells were also restored by upregulating PD-L1. In addition, the above cellular results were validated by the following in vivo results, and the results showed that overexpression of PD-L1 increased Cyclin D1 and CDK2 expression levels, while inhibited cleaved Caspase-3 and Bax expressions in the tumor tissues collected from xenograft mice administered with DB-Cisplatin combined treatment (P < 0.05, Fig. 6J-M).
{kind=link}
{kind=link}