Middle aged eNOS-deficient mice display severe myelin loss and axonal pathology
In our previous work, we characterized AD-like pathologies in eNOS-deficient mice at old age [16]. We have recently focused on characterizing middle-aged mice, since metabolic vascular factors pose the highest risk in this age group [5–7]. As reported, eNOS-deficient mice manifested most of the phenotypes of metabolic syndrome such as hypertension, insulin resistance, hyperlipidemia, hypertension, and hyperhomocysteinemia [28–30]. We now determined white matter changes at three ages (7 MO, 12 MO, and 24 MO) in eNOS-deficient (eNOS−/−) and littermate control mice (eNOS+/+). Coronal sections encompassing the brain regions corresponding from frontal to temporal cortical areas [+ 2.0 mm to -2.5 mm] were systematically examined by histological means for the expressional changes in axonal and myelin protein markers. Firstly, myelin integrity was assessed in eNOS mouse brains by both LFB (luxol fast blue) staining (Fig. 1A) and immunohistochemistry on myelin basic protein (MBP) (Fig. 1B). Significant myelin loss was first detected in the cortex and corpus callosum (CC), but not in the striatum in eNOS−/− mice at 12 months of age (12 MO) (Fig. 1 and SFigure 1). Myelin loss was also detected in eNOS heterozygous mice at mid-age (SFigure 1B) and thinning of the CC region, the largest white matter zone, was detected at old age (24 MO).
Surprisingly, Western blot data based on forebrain tissue lysates showed a nearly 2-fold increase in MBP protein in eNOS−/− at mid-age (12 MO) on all four isoforms from alternative splicing, with 17 kDa and 21.5 kDa being the predominant species (SFigure 1C) which presumably play differential roles of functional significance in oligodendrocytes. Notably, there was markedly increased colocalization of MBP immunosignals in the nuclei of eNOS−/− brain sections (SFigure 1D). It is unclear if these nucleus-translocated MBP species represent mis-aggregated dysfunctional protein which warrants further investigation via biochemical analysis. Nuclear MBP has been reported to display different roles in myelination compared to cytoplasmic protein species, most often seen with the 17 kDa and 21.5 kDa isoforms [31], appearing to be associated with the process during oligodendrocyte maturation before compact myelin formation [32]. Indeed, we observed similar results on two additional myelin protein markers Olig1 (oligodendrocyte transcription factor 1) and CC-1 (mature oligodendrocyte somata marker) (SFigure 2A and 2B), indicating the impaired function of myelin-producing oligodendrocytes which was supported by additional data to be presented later.
Severe gait imbalance and cortical pyramidal layer neurodegeneration were detected in middle aged eNOS-deficient mice
Cortical white matter change is often associated with gait dysfunction. We used the CatWalk system to assess gait performance of eNOS littermate mice in all three genotypes at 12 months of age. The test started once the mouse entered the visual field of the camera and stopped once the mouse disappeared. The averaged running speed is therefore reciprocal to the duration of the trial. Since the traces of each limb are marked, the moving speed can be alternatively calculated based on each limb. The immediate speed can be constant or vary heavily, so the variability is evaluated as well. We present data in Fig. 2A that the averaged running speed was consistently reduced in eNOS+/− mice and was even slower in eNOS−/− mice. Of note, eNOS−/− mice not only moved slowly, but also failed to run at a constant speed compared to eNOS+/+ and eNOS+/− mice. Moreover, eNOS-deficient mice (both eNOS+/− and eNOS−/−) displayed significantly altered patterns of gait sequence inter-limb coordination compared to eNOS+/+ mice. The same cohort of eNOS-deficient mice displayed significant deficits in two additional tests of rotarod and grip strength for motor functions at mid-age compared to eNOS+/+ mice (data not shown).
Cortical white matter demyelination predictably leads to cortical circuit dysfunction. We then examined the overall health of the neurons in both cortical and hippocampal regions of eNOS−/− and control mice by LFB-Nissl double staining. Strikingly we detected disorganized cortical structure with signs of severe neurodegeneration in cortical pyramidal neuronal layers II/III and V/VI at 12 months of age, revealing the presence of an empty vacuole surrounding the seemingly degenerated neurons due to loss of myelin sheath (Fig. 2B). Of note, we did not detect apparent neurodegeneration of hippocampal neurons at this age. Consistently, we detected a significant increase in the expression of activated caspase-3 in the forebrain samples of eNOS−/− mice compared to control littermate eNOS+/+ forebrain (SFigure 2C). Notably, myelin loss appeared to occur before the cortical pyramidal neurodegeneration since no neurodegeneration was detected at 7 months of age while there was a sign of demyelination (SFigure 3).
Elevated ROS and astrogliosis and neuroinflammation detected selectively in the white matter before the middle age
Since myelin-producing oligodendrocytes are the most energy-demanding cell type in CNS at the highest metabolic rate, they are known to be most vulnerable to stress such as ROS and hypoxia/ischemia. Indeed, we observed markedly elevated ROS levels by DHE staining, selectively in the parietal cortical region (Fig. 3A); no significant increase in DHE staining signal in the CC or striatal brain regions at middle age (SFigure 4A).
Furthermore, we found markedly increased fibrous astrocytes based on the expression of the glial fibrillary acidic protein (GFAP) in the CC region in middle-aged eNOS-deficient mice; they were aligned in rows between the axon bundles in parallel which displayed smooth and long processes of a fingerlike shape (Fig. 3B). Heterozygous eNOS+/− brains also displayed an increase in GFAP positive cells at 12 months of age, but at lesser degrees than eNOS−/− brains (SFigure 4B). Surprisingly, there was no significant increase in Iba-1 positive microglia in the brains of 12 months-old eNOS-deficient mice; a significant global increase was found at 24 months of age (Fig. 4C). It remains to be determined the precise order of these events at molecular and cellular levels.
Although GFAP is the main astrocytic intermediate filament, immunohistochemistry using multiple GFAP antibodies only labeled astrocytes in the corpus callosum, cerebral peduncle, and the hippocampus in mouse brain under physiological condition. Owning to the increasingly recognized heterogeneity of astrocytes from different brain regions [33], we used a second marker S100b which is more suitable for capturing the overall distribution of astrocytes in all brain regions. Indeed, we observed more S100b-positive astrocytes in the cortical area in eNOS−/− brain at mid-age; not only the number but also the fluorescent intensity of each astrocyte was markedly increased (SFigure 4C). Most strikingly, contrary to the globally increases S100b-positive astrocytes in both the CC and cortical areas in eNOS−/− brains, the GFAP-positive astrocytes were selectively found around the cortical layer II/III and V/VI regions, surrounding the degenerating/degenerated pyramidal neurons (SFigure 4D). Astrocytes are best known for multi-faces, in tissue repair, and in mediating an inflammatory response. Indeed, at higher magnification, both the GFAP- and S100b-positive astrocytes displayed thicker cellular process, indicating reactive astrocytes upon injury (SFigure4C and 4D).
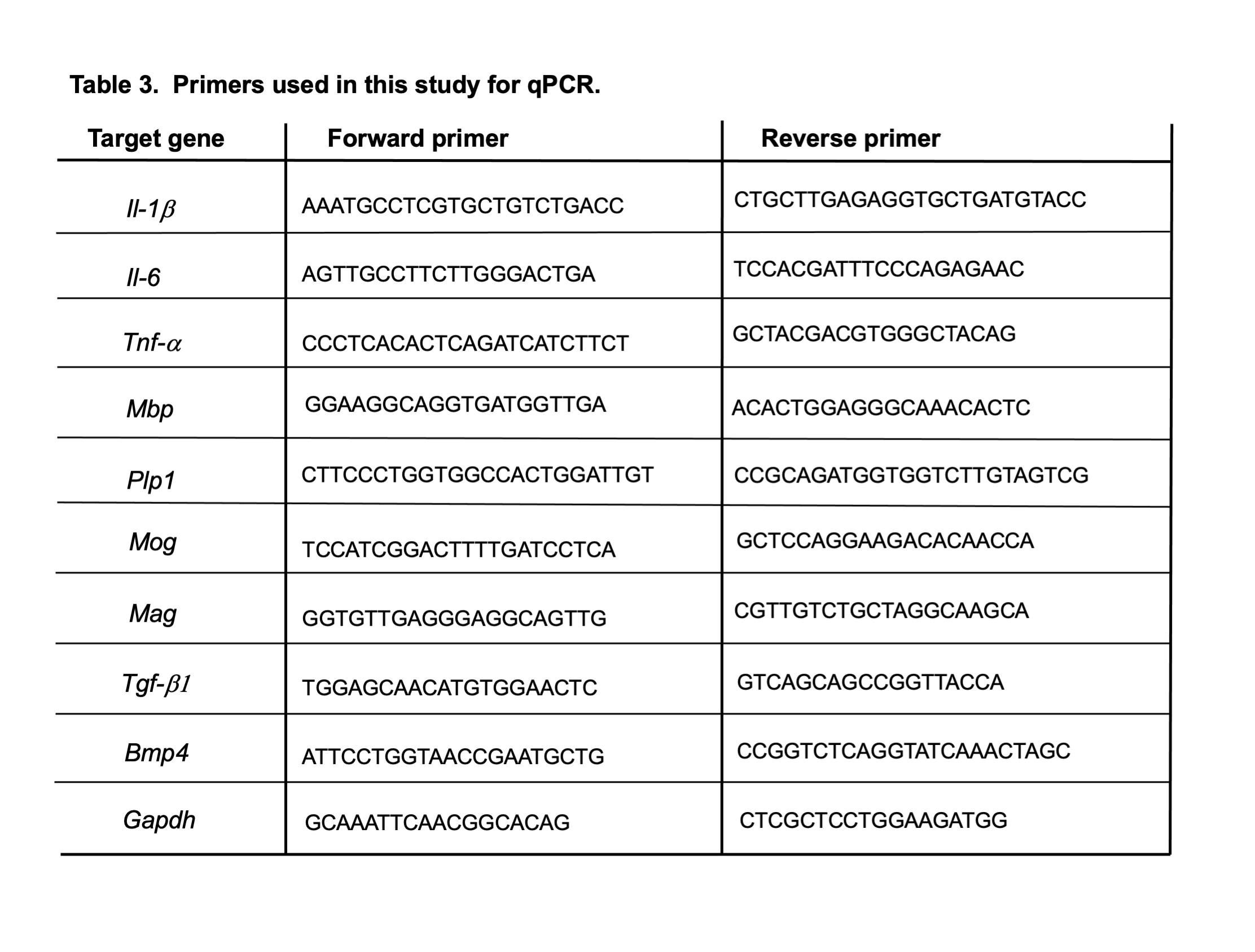
To verify that these newly increased astrocytes are indeed reactive astrocytes upon injury, we undertook a different approach of investigating cytokine gene expressional changes from the largest CC region (white matter, WM) and the adjacent subcortical region (gray matter, GM) (Fig. 4A). Most strikingly, markedly upregulated levels of three pro-inflammatory cytokine genes were detected only in the white matter of the eNOS−/− brain at 10 months of age (Fig. 4B). Because we did not observe increased microgliosis at this age, we reasoned that these cytokines were most likely released from the activated reactive GFAP-positive astrocytes. Given the sparse nature of the newly emerged GFAP-astrocytes in the specific regions of cortical areas in eNOS−/− mouse brains by mid-age, the upregulated inflammatory gene expression in gray matter may be oversight from the micro samples taken via micro punch which consisted of a majority of dense neuronal bodies (somas). Nevertheless, the prominently elevated cytokines in the CC region are the unambiguous evidence for neuroinflammation, specifically white matter neuroinflammation, as an early contributing mechanism to the cortical pathology such as demyelination and selective cortical neurodegeneration. Consistently, all four major myelin pathway genes (i.e., Mbp, Plp1, Mog and Mag) were found to be markedly downregulated in the CC region in eNOS−/− mice at 10 months of age as compared to littermate control mice based on quantitative qPCR data (Fig. 4C). The implication of impaired integrity of the myelinated axons and white matter axon-glial coupling upon eNOS deficiency will be discussed later.
Hypoperfusion and BBB leakage are the two earliest pathologies detected in eNOS-deficient mice
Endothelium-derived NO (EDNO) signaling has long been known to be crucial for angiogenesis and vasculogenesis during the early developmental stage. Using FITC-angiography, we detected reduced vessels from a dorsal view of eNOS-deficient mice at a young age (3 MO) (Fig. 5A, left two panels), which was similarly reported previously not attributable to impaired collateral formation in the embryo-neonate stage [34]. We therefore reasoned that this phenomena indicated arterial occlusion resulting from impaired NO signaling. Indeed, as we reported previously [16], regions of the hypoperfused brain and microvascular occlusion appear in eNOS-deficient mice at a young age, with markedly increased frequency of similar lesions in an age-dependent manner. As shown in a representative cortical section from young eNOS-deficient mice (3 MO) (the right panel of Fig. 5A), these lesions/occlusions are typically small (ranging from 100 to 200 µm in diameter); by middle age, they are frequently found bilaterally in the parietal association cortex (Table 1). This pathological pattern is highly reminiscent of the bilateral temporoparietal hypoperfusion characteristic of AD patients. Occlusions were not uniformly located throughout the eNOS−/− brain, but rather in defined areas, listed in order of higher to lower frequency: parietal association, temporal association and retrosplenial granular cortices, hippocampus, and thalamus. No such lesions were detected in the WT eNOS littermates up to 24 months of age.
In our prior work we also reported compromised BBB in middle to old-aged eNOS-deficient mice based on FITC-dextran (150 kDa) angiography as well as quantitative mouse IgG extravasation [16]. We speculated that BBB integrity and function may be compromised at a much younger age in eNOS-deficient mice owning to the indispensable role of EDNO signaling in endothelial vascular permeability via modulating VEGF signaling. To detect early BBB leakage, we systematically tested various molecular tracers reportedly used for this purpose including FITC-dextran species at different molecular weights and Evans blue dye. Surprisingly, when we used FITC-dextran of a lower molecular weight range (3–30 kDa) as a tracer, fluorescence was not detected in the brain, presumably due to rapid systemic clearance. In the end, we concluded that Evans blue dye is an optimal fluorescent tracer for the degrees of leakage in the eNOS model.
Evans blue (980 Da) is an azo dye with a very high affinity for serum albumin (67 kDa). Because serum albumin cannot cross the BBB and virtually all Evans blue is bound to albumin, albumin-bound Evans blue enters the parenchymal tissue in CNS when the BBB has been compromised. Since Evans blue fluoresces with excitation peaks at 470 and 540 nm and an emission peak at 680 nm, it can be detected in brain sections under a fluorescent microscope using a Rhodamine filter. After injecting Evans blue dye to mouse tail vein, the brains were removed shortly and post-fixed and processed to serial 100 mm coronal sections for angiographic examination. By this method, we detected massive BBB leakage in eNOS−/− mice beginning at a very young age (6–8 week-old). As shown in Fig. 5B, eNOS-deficient brain at 3 months of age displayed numerous diffusive spots indicating BBB leakages spanning from frontal to parietal cortical regions compared to no leakage found in littermate control mice. The gradually decreased leakage spots in the five representative angiographs spanning Bregma + 2.0 mm to -1.82 mm indicates that leakages start predominantly from an anterior to posterior direction. Indeed, we detected more severe leakage expanded to deeper and posterior areas in older eNOS-deficient mice. Strikingly, like FITC-non-perfusion lesions, there was no leakage detected in wild-type eNOS mice even at 24 months of age using this methodology (Table 1). Interestingly, we examined mouse brain sections after co-injection of FITC-dextran (2000 kDa) and Evans blue dye and found largely non- overlapping fluorescent signals in eNOS−/− brain at a young age (data not shown), suggesting that these two early pathological events of non-perfusion and BBB leakage are independent of each other, both resulting from the impaired EDNO signaling.
Accordingly, we detected massive upregulation of metalloproteinases 9 (MMP9) at middle age by both immunohistochemistry and Western blot analysis, with MMP9-positive signals largely colocalizing with the Evans blue leakage signals (SFigure 5A-C), indicating breakdown of the endothelial basement membrane. Surprisingly, we detected no significant changes in MMP2 expression, and laminin expression was only elevated at 24 months of age (SFigure 5D). Since astrocytes contact blood capillaries with their endfeet as an integral part of the BBB interface, they are crucial in governing and maintaining water homeostasis in the CNS. We therefore also investigated aquaporin-4/Aqua4, a marker of astrocytic endfeet surrounding the capillaries by immunohistochemistry. Aqua4 proteins displayed more intense expression in microvessels in gray matter than in white matter, consistently because the microvessel density is much lower in white matter. Although Western blot data showed no significant change in Aqua4 protein expression levels from forebrain tissues of eNOS−/− mice at 12 months of age, Aqua4 proteins appeared to undergo redistribution upon eNOS deficiency by middle age; there were slightly more intense Aqua4 immunosignals overlapping with GFAP-positive astrocyte endfeet along the microvessels (SFigure 6B), suggesting altered BBB functions. Western blot analysis revealed nearly complete loss of Aqua4 protein expression in old eNOS−/− mouse brain at 24 months of age (SFigure 6C). Similarly, we detected a significant reduction of glucose transporter 1/GLUT1 expression only at an older age (SFigure 6D).
Restoration of NO signaling via sodium nitrate (SN) feeding rescues white matter pathologies in eNOS-deficient mice
Restoration of NO signaling with 1 mM SN in drinking water for up to 10 weeks has been reported to reverse hypertension, systemic hyperlipidemia, and glucose intolerance in 14- to 22-month-old eNOS−/− mice [27]. This prompted us to use this agent to test our hypothesis that feeding eNOS-deficient mice with SN in drinking water at a very young age, starting from 6-week-old for 6 weeks, would prevent the development of the two early pathological events namely hypoperfusion and BBB leakage. Strikingly, 6-week SN feeding completely prevented BBB leakage and non-perfusion lesions in eNOS−/− mice (Fig. 5C and Fig. 6B and 6C Cohort 1 mice; n = 3/group). These promising data encouraged us to further testing the possibility of chronic feeding sodium nitrate in drinking water to eNOS-deficient mice at older post-symptomatic age.
In addition to the first cohort of mice that received 6-week-SN feeding which resulted in nearly complete prevention of BBB leakage (Fig. 5C and Fig. 6B), we also fed mice for a longer period in eNOS−/− mice for over 6 months (Cohort 2) and post-symptomatically (Cohort 3, 11–16 months) (Fig. 6A). We focused on ROS, white matter inflammation, and myelin preservation in the Cohort 2 mice. Specifically, SN feeding completed prevented pro-inflammatory cytokine gene upregulation in eNOS-/- white matter tissue at 10 months of age (Fig. 4B) and the decline of myelin pathway gene expression (Fig. 4C). It also prevented ROS elevation and astrogliosis in selective cortical layers (data not shown). Furthermore, loss of myelin neurofilaments (SMI 132) in eNOS−/− brains was significantly rescued by SN feeding (Fig. 6C). Lastly, gait performance was significantly improved in Cohort 3 mice when tested at 16 months of old age, after SN feeding was initiated from 11 months of age in eNOS−/− mice (Fig. 6D). Although the SN eNOS−/− mice could not perform as well as the eNOS+/+ mice at this old age (16 MO), this result is most encouraging with translational value for a potential reversal effect of cortical neuronal connectivity and functions. Taken together, chronic SN feeding resulted in significant prevention or improvement in white matter pathologies and function in eNOS−/− mice.
{kind=link}
{kind=link}
{kind=link}