Patients and tissue samples
Patients enrolled in this study were independently diagnosed with primary glioma by two pathologists in a double-blinded manner according to the criteria of the 2016 WHO classification. They had undergone routine surgery at the Department of Neurosurgery of Nanfang Hospital (Guangzhou City, Guangdong Province, China) between 2013 and 2019 without radiotherapy or chemotherapy prior to surgery. Brain tissues beyond MRI indicated peritumor edema area were collected as peritumor tissues. Temporal tissue samples from 3 epilepsy patients undergoing surgical treatment were used as normal brain tissues. This study was permitted by the Ethics Committee of Southern Medical University, and informed consent was obtained from each of the enrolled patients.
Cell lines and culture
Human glioma cell lines U87MG, T98G, LN229, and SVG p12 were purchased from American Type Culture Collection (ATCC: Rockville, MD, USA). The primary human glioblastoma cell line NFHDCD was derived and cultured from a patient pathologically diagnosed with GBM (Male, Aged 60, GBM, WHO IV, IDH+, MGMT+, TP53+, Ki67+, EGFR+) who underwent routine surgery at the Department of Neurosurgery of Nanfang Hospital (Guangzhou City, Guangdong Province, China)[24]. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM glucose 4.5 g/L; Biological Industries) with 10% fetal bovine serum (FBS; Biological Industries) at 37 °C in a humidified incubator with 5% CO2.
Immunohistochemistry (IHC)
Seventy-five paired human glioma tumor and peritumor tissues were used for immunohistochemistry experiments to study altered TMEFF2 protein expression using the two-step plus poly-horseradish peroxidase (HRP) method. The clinical information of the studied specimens was shown in Table S1. Briefly, 4 μm sections were mounted on amino propyl ethoxysilane (APES) slides. The slides were deparaffinized, rehydrated, immersed in 10 mM sodium citrate buffer (pH 6.0), pretreated in a microwave oven for 20 min, and then rinsed for 15 min with phosphate-buffered saline (PBS). Endogenous peroxidase was quenched by incubation of the sections in 0.3% hydrogen peroxide for 30 min at room temperature. Nonspecific binding was blocked by incubation with nonimmune serum (1% bovine serum albumin for 15 min at room temperature). The sections were incubated overnight with two polyclonal antibodies against TMEFF2 (rabbit anti-TMEFF2, ab50002, Abcam, Cambridge, UK; and rabbit anti-TMEFF2, 11928-1-AP, Proteintech, Rosemont, IL, USA) at a dilution of 1:1000. The next day, the slides were stained with a two-step plus Poly-HRP Anti-Rabbit IgG Detection System (PV-6001; ZSGB-Bio, Beijing, China) to detect TMEFF2. After visualization of the reaction with 3,3′-diaminobenzidine, the slides were counterstained with hematoxylin and mounted with synthetic medium. Gastric cancer tissue was used as a positive control, and PBS replaced the anti-TMEFF2 primary antibody to provide a negative control condition.
RNA isolation and qRT-PCR
Twenty-three paired human glioma tumor and peritumor tissues were used in qRT-PCR experiments to detect TMEFF2 mRNA expression. The clinical information of the studied specimens was shown in Table S2. RNA isolation and qRT-PCR were carried out as previously described[25]. Total RNA was isolated from twenty-three paired glioma tumor and peritumor tissues as well as LN229, T98G, U87MG, and NFHDCD cells using TRIzol (Invitrogen), and RNA samples (600 ng per sample) were used to generate cDNA using a PrimeScript™ RT reagent Kit with gDNA Eraser (Takara, Cat# RR047A) according to the manufacturers’ instructions. The obtained cDNA samples were used as templates for qPCR amplifications using TB Green® Premix Ex Taq™ II (Tli RNaseH Plus) (Takara, Cat# RR820A). GAPDH was used as a corresponding internal control. All mRNA levels were quantified by the 2-ΔΔCT method. Each reaction was performed in triplicate. The primer sequences of TMEFF2 were as follows: forward 5’-GCTGCTTTCCCTACCTCCTT-3’; reverse 5’-AGCCACACACAGGCACATAG-3’. The primer sequences of GAPDH were as follows: forward 5’-TGACTTCAACAGCGACACCCA-3’; reverse 5’-CACCCTGTTGCTGTAGCCAAA-3’. The primer sequences of DNMT1 were as follows: forward 5’- GATCTCCTACAACGGGGAGC-3’; reverse 5’- AGCCACCAATGCACTCATGT-3’.
DNA bisulfite conversion and methylation analysis
A DNA extraction kit (Takara, Cat# 9765) was used to isolate DNA from GBM and SVG p12 cells according to the manufacturer’s instructions. Sodium bisulfite conversion of 600 ng of extracted DNA was performed using a DNA Bisulfite Conversion Kit (Tiangen, Cat# DP215-02). Bisulfite conversion was followed by bisulfite amplicon sequencing (BSAS) or methylation-specific PCR (MSP). Bisulfite amplicon sequencing (BSAS) was performed within CpG sites of the promoter region of TMEFF2. The four primers that were used to amplify TMEFF2 are shown in Table S3. Methylation data were then analyzed, and the average methylation levels at all sites were calculated using MethylKIT software (Access Date 2019/09/26). The methylation level of each CpG site is defined as the ratio of the number of methylated reads to the combination of methylated and unmethylated reads (values between 0 and 1). For methylation-specific PCR, an EpiScope MSP Kit (Takara, Cat# RR100A) was used, and qRT-PCR was performed. The primers specific for TMEFF2 in MSP are shown in Table S3. For MSP of clinical glioma samples, tumor tissues from forty-three patients who had undergone whole exome sequencing in the clinic, which determined the mutation status of the IDH1, ATRX and TP53 genes, were used. The clinical information of the studied specimens was shown in Table S4. Temporal tissue samples from 3 epilepsy patients undergoing surgical treatment were used as normal controls.
5-Aza-2-deoxycytidine decitabine (DAC) treatment
DAC was purchased from Selleck Chemical Co. (Selleck, Cat# S1200) and was dissolved in dimethyl sulfoxide (DMSO) (Sigma, Cat# D2650). The stock solution was diluted with PBS to an original concentration of 10 mM and stored at -20℃. Further working solutions were added to the cell culture medium immediately before use. Appropriate DMSO controls were implemented. Cells were treated with DAC at a concentration of 5 μM for 96 h, after which total RNA or DNA was extracted from the cells.
Transient knockdown of DNMT1 and TMEFF2 in GBM cells
Cells were transfected with chemosynthetized siRNAs purchased from Kidan Biotechnology Co. (Guangzhou, China) using Lipofectamine 2000 reagent (Invitrogen, Cat# 11668) for 8 h according to the manufacturer’s protocol. The sequences of the siRNAs are shown in Table S3.
Cell proliferation assay
Cell proliferation was measured by Cell Counting Kit-8 (CCK8) assays and EdU assays.
For CCK8 assays, cells were seeded in 96-well plates at a density of 2000 cells/well and then were incubated for 12 h to allow cell attachment. Then, 10 μL of CCK-8 solution (Bimake, Cat# B34304) was added to each well on days 1, 2, 3, 4, 5 and 6, which was followed by another 2 h incubation. The optical density was then measured at 450 nm using a microplate reader.
For EdU assays, cells were plated at a density of 20000/dish in confocal dishes. After 24 h of incubation, cells were treated with EdU reagent (RiboBio, Cat# C10310-1) for 2 h according to the manufacturer’s instructions and then were fixed with 4% paraformaldehyde. One hundred microliters of 1X Apollo®567 staining reaction solution and Hoechst 33342 (RiboBio, Cat# C10310-1) was added to each dish and then was incubated for 30 min at room temperature on a decolorization shaker. Cells were then visualized using a BX63 automatic intelligent fluorescence microscope (Olympus, Tokyo, Japan).
Database and data analysis
Validation cohort data was collected from the GBM and LGG projects of The Cancer Genome Atlas (TCGA). DNA Methylation (Illumina 450K), Mutations and Clinical Data sets were downloaded from the cBioPortal database[26], and RNA HiSeq V2 RSEM data were downloaded from the GDC Data Portal[27]. RNA HiSeq V2 RSEM data were transformed from FPKM into TPM data before merging into the matrix.
Statistical and survival analysis
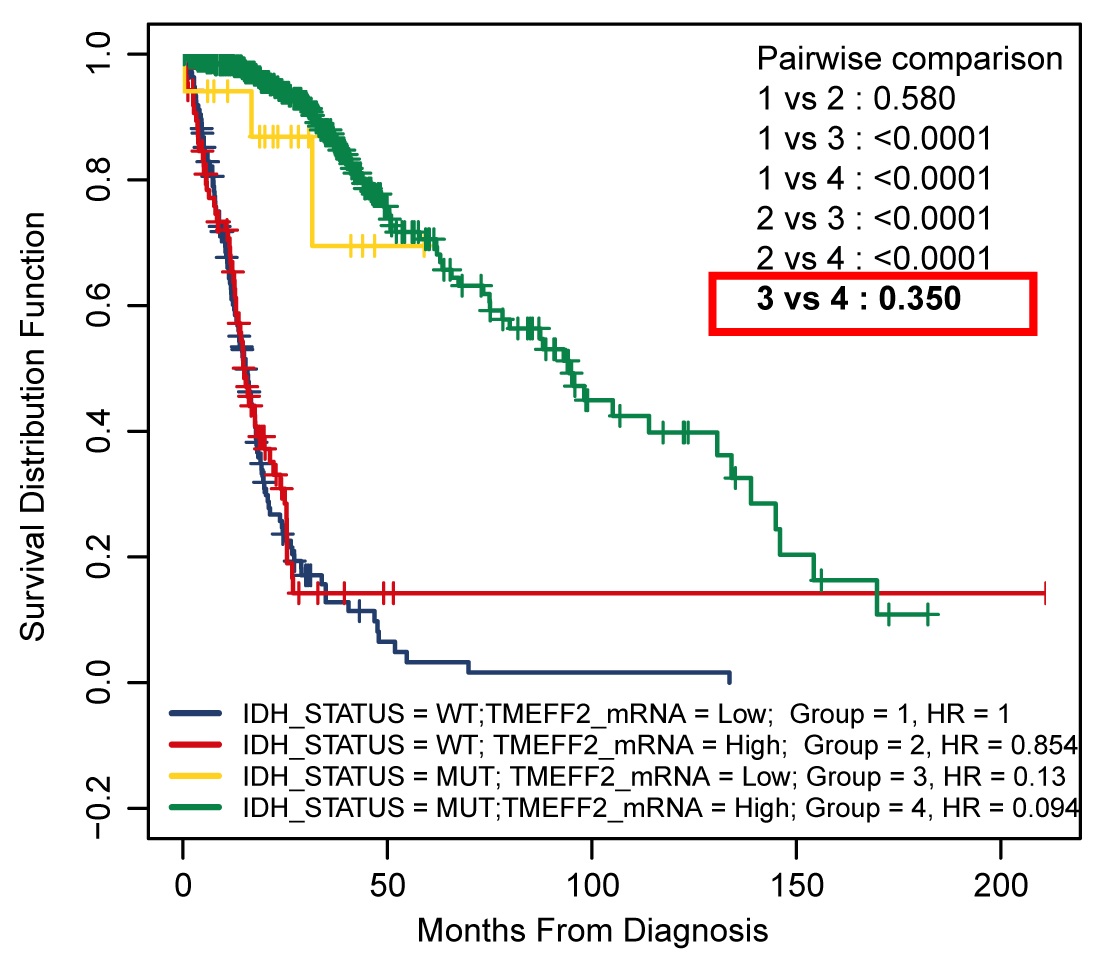
R software version 3.5.0 was used to assess the relationship between the TMEFF2 methylation level and mRNA expression through the use of Spearman’s correlation analysis. Kruskal-Wallis tests were used to analyze TMEFF2 methylation or mRNA expression in different cells as well as in different tumor grades. The Wilcox test was used to analyze the correlation between TMEFF2 methylation or mRNA expression and gene mutations. Kaplan-Meier analysis was used to assess the association of TMEFF2 methylation or TMEFF2 mRNA expression with patient prognosis. A scanning model found that β value = 0.1 was the best cutoff for TMEFF2 methylation and that TPM = 6.07 was optimal for assessing TMEFF2 mRNA expression in Kaplan-Meier analysis of the TCGA cohort. Differences between survival rates were analyzed using a log-rank test, and survival curves were plotted using R software. Differences in TMEFF2 methylation and expression in different treatment groups were analyzed using Student’s t test, and CCK8 results were analyzed using variance analysis of two-factor repeated measures. Statistical analyses were carried out using SPSS statistical software (version 20.0, SPSS, Inc., Chicago, IL, USA), and the significance level was assigned at P <0.05.
{kind=link}