Cell culture
Human breast cancer cell lines MCF-7 and MDA-MB-231(obtained from ATCC) were grown in DMEM (GIBCO) containing 10% FBS (GIBCO). The cell lines were maintained at 37°C in a 5% CO2 incubator.
MTT Assay
MCF-7, MDA-MB-231, and MCF-7 SHDDR1 were separately cultured in 10% FBS growth medium in 96-well plates (5000 cells/well) overnight. The cells were treated with various concentrations of Nilotinib (n=11; 14.5-30,000 nM) and cultured in 10% FBS medium for 48 h in triplicates. The control cells were treated with dimethyl sulfoxide (DMSO) only. Next, tetrazolium dye (MTT) solution (5 mg/mL, 20 µL/well) was added to each well. After incubation for 4 h at 37°C, the supernatant was aspirated, and the generated formazan crystals were dissolved in 150 µL of DMSO, and the absorbance was recorded spectrophotometrically at 490 nm using an enzyme-linked immunosorbent assay plate reader. The data were calculated using Graph Pad Prism version 5.0. The fitting of the IC50 values was done using a non-linear regression model with a sigmoidal dose-response.
Cell Apoptosis Assay
MCF-7 and MDA-MB-231 (3 × 105) cells were seeded into 6-well plates overnight. Fresh growth media with Nilotinib (500 nM) as well as medium with 1‰ DMSO (control) was added to the plates. After 48 h, the growth medium was collected, and cells were trypsinized and collected with the corresponding medium. After centrifugation at 1000 × g at 4 ºC for 3 min, the supernatant was removed completely, and the cells were washed twice with cold PBS. Then, 100 µL of 1× binding buffer, 5 µL PI (PI, BD) and 5 µL annexin-V (FITC-Annexin V, BD) were added. The cells were then gently vortex-mixed and incubated for 15 min at room temperature in the dark and 1× binding buffer was used for dilution to 500 µL. The cells were then stained with PI, and Annexin-V alone as a positive control. The samples were measured using a BD Accuri™ C6 flow cytometer (Becton Dickinson), and the data were processed using FlowJo 7.6.1.
Western blotting
Protein extracts were obtained using the KEYGEN total protein extraction kit (Nanjing, China). The concentration of protein in the supernatant fractions were determined using a BCA Protein Assay Kit (Pierce). Sixty micrograms of protein per sample were loaded and separated on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE), followed by electrophoretic transfer onto a polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA, USA). After blocking with 5% skim milk in Tris-buffered saline containing 0.05% Tween-20, the membrane was first incubated with the indicated primary antibody at 4°C overnight. Signals were detected using an ECL+TM Western blotting system (Bio-Rad, Hercules, CA). Primary antibodies used were: antiphospho-DDR1 (cst#11994), anti-DDR1, anti-ERK1/2, antiphospho-ERK,E-Cadherin, Vimentin, Snail 1, and ant-GAPDH (Cell Signaling Technology).
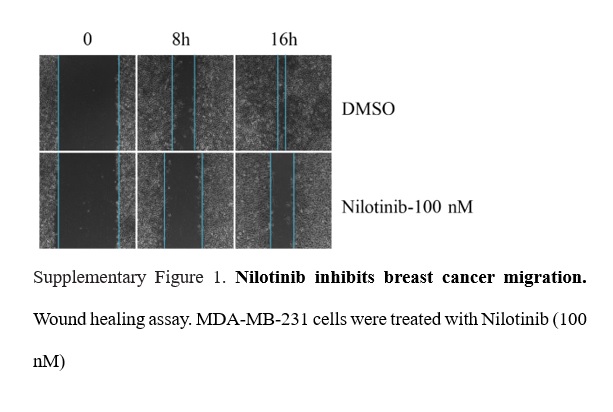
Wound-healing assay
MCF-7 SHctrl and MCF-7 SHDDR1 cells were seeded in 6-well plates and incubated at 37°C overnight. After reaching 100% confluence, a straight line of similar width was scratched across the monolayer using a 10-μL pipette tip, for each sample. After washing with PBS to remove non-adherent cells, cells were then treated with the indicated concentrations of Nilotinib and incubated for 24 h. When the wound in the DMSO control was healed, the image from the original scratch line was captured by Leica DFC 3000 G.
Immunofluorescence staining
Cells were seeded in a confocal dish, fixed with ice-cold 4% paraformaldehyde and incubated with 0.5% bovine serum albumin in phosphate-buffered saline (PBS) with 0.1% Triton X-100 for 30 min. After washing, cells were incubated with anti-DDR1 antibody before rinsing with PBS, then immunostained with secondary antibodies and stained with 4′,6-diamidino-2-phenylindole (DAPI) (2 mg/ml). Fluorescence images were captured using a confocal microscope (Leica TCS SP2, Germany).
Lentiviral production and transduction
For viral creation, DDR1 shRNA was designed based on the DDR1 mRNA sequence (GenBank accession no. NM-001954), the shrna#1 sequence was CCTATACGTTTCTGTGGAGTA, and shrna#2 sequence was TGCTGACATGAAGGGACATTT. Each of these sequences were cloned into a plko.1 lentiviral vector. The digestion analysis of restriction endonuclease confirmed the recombinant vector, and all inserted sequences were verified by DNA sequencing. Lentiviruses were developed by triple transfection of 80% confluent 293T cells with DDR1 shRNA-expressing vector and the virion-packaging elements (pVSVG-I and pCMVΔR8.92) using Lipofectamine 2000 (Invitrogen). They were harvested in a serum-free medium after 48 hours and filtered through a 0.45mm filter (Millipore, Bedford, MA). Retroviruses harboring shRNA sequence were transduced into MCF-7. After incubation for 48 hours, the transduced cells were positively selected in puromycin (1μg/mL), passaged, harvested, and named MCF-7 SHDDR1#1 and MCF-7 SHDDR1#2. And control shRNA lentiviral particles (obtained from Santa Cruz Biotechnology).
Statistical analysis
Statistical analyses were conducted using GraphPad Prism 5 software. Differences were analyzed using one-way analysis of variance followed by Tukey's post hoc test for multiple comparisons and Student's t-test for two comparisons. Data were presented as the means ± SD. *, ** and *** indicates P < 0.05, P < 0.01 and P < 0.001, respectively.
{kind=link}