Isolation, purification and culture of bovine trophoblast cells
Placentae of early pregnant Holsteins (45–60 days of pregnancy, with a fetal cow with a crown-to-rump length of about 7 cm) were obtained from the cows sacrificed by exsanguination. This study was performed in accordance with the guidelines of the Animal Ethic Committee of Beijing University of Agriculture (Permit No.: SYXK(JING)2015-0004). The isolation of cells was performed according to the previous study[6]. Briefly speaking, the fetal cotyledons were separated from the maternal caruncles aseptically and minced into 1mm3 pieces, which were dispersed into a 10 cm2 dish and cultured in a 37 oC incubator for 30 min till the pieces conglutinated on the dish sturdily. The pieces were rinsed and cultured with complete DMEM/F12 medium (Gibco, USA) supplemented with 10% exosome-depleted fetal bovine serum (FBS) (EXO-FBS-50A-1, SBI, USA) at 37oC in an atmosphere of 5% CO2. The medium was changed every 3 days till the cells can be seen under a phase-contrast invert microscope. The cells were purified with the differential velocity adherent method[24]. Immunofluorescence detection was used to identify the primary bovine trophoblast cells (described below).
Giemsa stain assay
The cells were fixed with 4% paraformaldehyde for 40 min and washed with PBS. After being dyed with Giemsa stain for 20 min, they were observed under a Nikon inverted optical microscope (Nikon, Japan).
Cell viability assay
According to the manufacturer’s instructions (Solarbio, Beijing, China), the cells were seeded in a 96-well plate at the density of 104 cells per well, incubated with CCK-8 solution for 1 h, and then measured for the absorbance using a wavelength of 450 nm. The relative cell viability was calculated with the following formula: Relative Cell Viability = (The Absorbance of Test Group / The Absorbance of Control Group) * 100%.
Enzyme-linked immunosorbent assay (ELISA)
Analysis of the endocrine ability of hormone was performed according to the manufacturer’s instructions (J༆L Biological, Shanghai, China).
Western blot
The cells or exosomes were lysed with RIPA buffer supplemented with phenylmethanesulfonyl fluoride (PMSF) for 30 min on ice. The lysates were centrifugated at 12,000 rpm for 20 min at 4 oC. The supernatants were collected and measured for the total protein concentration with a BCA kit (Beyotime, Beijing, China). The lysates of cells or exosomes were separated on SDS-polyacrylamide electrophoresis gels (4% stacking gel and 12% resolving gel) and transferred onto PVDF membranes (Millipore, Massachusetts, USA) using an electroblotting apparatus. Then the membranes were blocked with 4% non-fat milk for 2 h and incubated with primary antibodies including CK7 (ab154334, 1:1,000, Abcam), vimentin (ab45939, 1:1,000, Abcam), E-Cadherin (ab40772, 1:1,000, Abcam), CD90 (ab92574, 1:1,000, Abcam), hTERT (ab32020, 1:1,000, Abcam), CD9 (orb94982, 1:1,000, Abcam), CD63 (ab134045, 1:1,000, Abcam), MUC 1 (1:1,000, Beijing Jiaxuan Biotech Co., Ltd.), integrin αv (1:1,000, Beijing Jiaxuan Biotech Co), integrin β3 (1:3,000; Beijing Jiaxuan Biotech Co., Ltd.), Wnt7a (ab100792, 1:3,000, Abcam), CK8 (ab53280, 1:1,000, Abcam) and GAPDH (bsm-33033M, 1:2,000, Bioss) at 4℃ overnight. They were washed with PBST and incubated with horseradish peroxidase (HRP) - conjugated goat anti-rabbit (bs-0295G-HRP, 1:3,000, Bioss) or goat anti-mouse (bs-0368G-HRP, 1:3,000, Bioss) secondary antibody for 2 h at room temperature. The proteins were visualized using an ECL chemiluminescence kit (Beyotime, Beijing, China). The densitometry of each immunoblot was performed using Image J software (National Institutes of Health, USA). With GAPDH as control, the expression of total protein was normalized.
qRT-PCR
Total RNA was isolated from the cells with TRIzol reagent (Invitrogen, USA). The concentration and quality of RNA was determined using an Agilent 2100 Analyzer and an NanoDrop 2000 microspectrophotometer. Complementary DNA was reversely transcribed from total RNA using HiFi-MmlV cDNA kit (CWBIO, Jiangsu, China) and qRT-PCR for gene expression was performed on an ABI 7500 Sequencing Detection System (Applied Biosystems, USA) with the UltraSYBR One Step qRT-PCR Kit (CWBIO, Jiangsu, China). The specific primers were designed as follows: 5’-TATGCCGTGGTCCAGAAGG − 3’ (hTERT, sense), 5’-CAAGAAATCATCCACCAAACG − 3’ (hTERT, antisense); 5’-CGGCACAGTCAAGGCAGAGAAC − 3’ (GAPDH, sense), and 5’-CCACATACTCAGCACCAGCATCAC − 3’ (GAPDH, antisense).
Immunofluorescence assay
The cells were fixed with 4% paraformaldehyde for 40 min and washed with PBS. The washed cells were immersed with permeabilization buffer for 30 min (PBS, 0.1% Triton X-100 and 1% BSA) and blocked with 1% BSA-PBS for 1h at room temperature. The cells then were incubated with primary antibodies including CK7 (1:100), vimentin (1:100), E-Cadherin (1:100), CD90 (1:100), CK8 (1:100) at 4 oC overnight, washed with PBS and incubated with fluorescein isothiocyanate (FITC) - conjugated goat anti- rabbit secondary antibody (ab6881, 1:100, Abcam) in the dark for 1h at 37℃. The cells were washed with PBS three times and incubated with DAPI for 5min at room temperature. Next, the cells were observed with a laser scanning confocal microscopy (Olympus, Japan) after being washed with PBS three times.
Soft agar assay
Soft agar assay was performed according to the published protocol[25]. The 1.2% agar was filled into a six-well plate as a bottom layer which then was placed still at 4 oC till the agar solidified. 5×104 cells were suspended in 1 mL 0.6% noble agar in DMEM/F12 media and plated onto wells. The cells were cultured at 37 oC in an atmosphere of 5% CO2 overnight and complete DMEM/F12 medium was added. They were fed with complete medium once a week and evaluated for the colony growth over the next two weeks. The cell clumps greater than 100 mm were considered as colonies and photographed with an inverted optical microscope.
Migration and invasion assay
The migration ability of BTCs was assessed using a 24-well plate in the BD Bio‐Coat Matrigel Invasion Chamber (BD Bios-ciences). 104 trypsinized cells were added into 1 mL serum‐free media. 500 µL complete media was added to the lower well while 100 µL cell suspension to the upper well. The plate was incubated at 37 oC in a 5% CO2 atmosphere for 24 h. Those non‐invading cells on the upper surface were removed using a cotton swabs and the lower surface of the basement membrane was fixed in 4% paraformaldehyde for 30 min, and then the basement was washed with PBS three times and stained with crystal violet. The cells were observed and photographed with an inverted optical microscope.
Cell transfection
The plasmid was transfected into the cells using lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s instructions. For transfection, the cells were seeded in 6-well culture plates and incubated with plasmid - lipofectamine complex at 50 nM in serum-free OPTI-MEM medium. The medium was replaced after 4 hours after transfection. The cells with 400mg / mL G418 (Solarbio, Beijing, China) were selected during two weeks. After selection, the transfected cells were subjected to the treatment for the follow-up experiment. pCI-neo-hTERT plasmid was identified and donated with Pro.Jin[22].
Exosomes isolation
To imitate the early embryo culture conditions, the cells were treated with 2 ng/mL P4 when their density was about 80%. After 48h, the medium was collected and centrifuged at 4 oC (300×g, 10 min to remove the cells), and then the supernatant was transferred to a sterile vessel. Exosome isolation were performed from the supernatant according to the manufacturer’s recommendations (BB-3901, Shanghai Bestbio Biotechnology Co., Ltd.).
Examination of exosomes morphology with transmission electron microscopy
The exosomes were resuspended in phosphate-buffered saline (PBS), and 15 µL samples were added to a 300-mesh copper grid for 90 s and dried. Each grid was washed five times in distilled H2O, stained with 5µL of 3% uranyl acetate (phosphotungstic acid) for 1min and dried, and then placed on a filter paper for 1h at room temperature. The samples were visualized with a transmission electron microscope at 80 kV.
Sample preparation for proteomic analysis
Lysates from the exosomes were extracted with lysis buffer (500 mM Tris-HCl, 50 mM EDTA, 700 mM sucrose, 100 mM KCl, 2% β-mercaptoethanol and 1 mM phenylmethylsulfonyl fluoride, pH 8.0), the protein was solidified by phenol-acetone assay and the precipitation was solubilized in 4% SDS. The protein concentrations were calculated by BCA assay and the enzymolysis protein concentrations tested by FASP assay.
DIA quantitation analysis
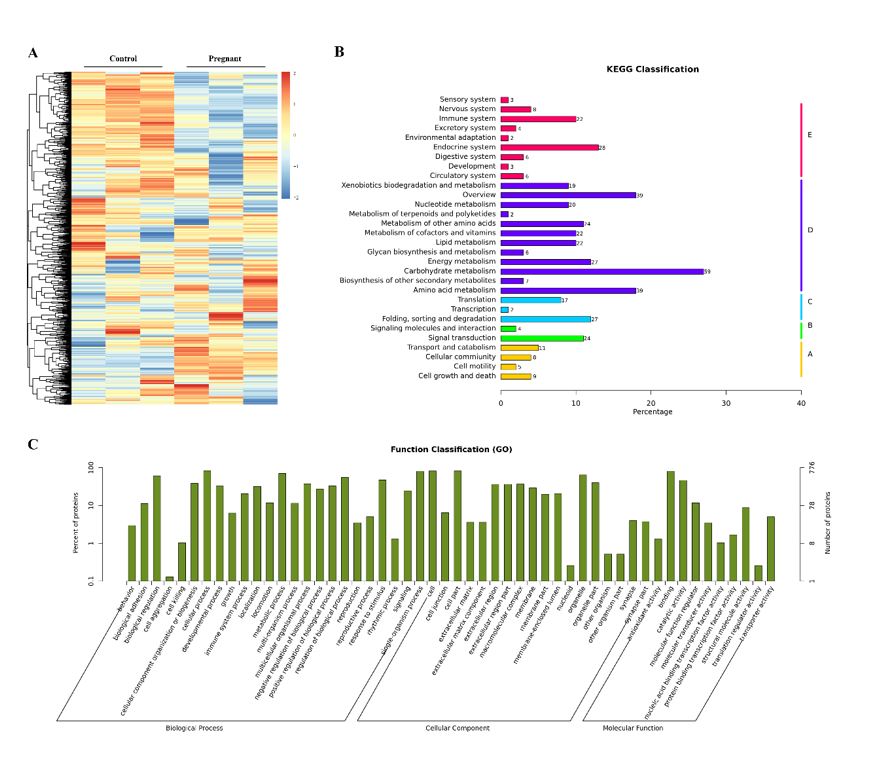
Proteomic experiments were performed in biological triplicate. MS analyses were performed on an Agilent 1100 high-performance liquid chromatography (Agilent, USA). Peptides were loaded and separated over a 120 min gradient run using a Waters XBridge C18 analytical column (5µm 120 Å, 460µm × 250 mm, Waters, China). Trapping was conducted for 3 min at 5µL min− 1, 97% buffer A (99% water, 2% ACN, pH 10.0), and 3% buffer B (98% ACN, 2% water, pH 10.0), before being eluted at 2-100% 0.1% FA in acetonitrile (2–40% from 0 to 100 min, 40–80% from 100 to 110 min (flow rate, 250 nL min− 1). According to the building requirements of the DDA library, after the flow-through peaks were removed, cross-merging, vacuum concentration and mass spectrometry library building analysis were completed. Proteome Discoverer 2.1 (Thermo Fisher Scientific, Rockford, IL, USA) was used to search and analyze the DDA data collected after classification. Biological samples were collected for DIA data, and quantitative analysis was performed with Skyline software (Department of Genome Sciences, University of Washington, Ave. NE, Seattle, WA). For pathway analyses, Kyoto Encyclopedia of Genes and Genomes (KEGG) resources were utilized with recommended analytical parameters. For gene ontology enrichment and network analyses, UniProt (www.uniprot.org) database resource (biological process, molecular function), Ingenuity Pathway Analysis, and Reactome knowledgebase were utilized. The heat map of proteins adopted gplots.
Statistical analysis
The research values were presented as mean ± SD. The data from three independent experiments were analyzed. One-way analysis of variance (ANOVA) was used to compare with the multiple time points and concentrations in the experiments. When the comparison between two groups means significant, * or + was used for P < 0.05, and ** or + + for P < 0.01.
{kind=link}