Clinical samples
For the IUA group, the endometria of six patients with IUA were collected. For the normal control group, fresh endometrial tissue was collected from six patients who underwent hysterectomy because of cervical intraepithelial neoplasia or subserosal fibroids or hysteroscopic endometrial biopsy due to infertility. The patients were 30-45 years of age and had regular menstrual cycles (25 d-32 d). No hormones or intrauterine devices (IUDs) were used by the patients during the three months prior to surgery, and postoperative pathology suggested no endometrial lesions. Endometrial extraction was reviewed and approved by the Ethics Committee of Zhujiang Hospital of Southern Medical University, and written informed consent was obtained from each patient prior to surgery. All presentations of case reports have consent for publication.
Immunohistochemistry
Tissues were fixed, paraffin-embedded and sectioned (5-μm continuous sections). H&E staining and Masson staining were performed. Paraffin sections were deparaffinized and rehydrated. The sections were heated in sodium citrate buffer in a microwave for antigen retrieval, washed with phosphate-buffered saline (PBS) for 5 min (three times) at room temperature, incubated in 3% H2O2 at room temperature for 25 min, washed with PBS for 5 min (three times) and blocked and incubated in goat serum for 30 min. The primary antibody was added dropwise (1:50, Abcam, Ab194427), and the sections were incubated in a 4 °C freezer overnight. The next day, the primary antibody was discarded, and the sections were washed with PBS for 5 min (three times). At 50 min after addition of the secondary antibody (1:200, SignalStain® Boost IHC Detection Reagent, CST, USA), the sections were washed with PBS for 5 min (three times). Finally, freshly prepared diaminobenzidine (DAB) was added for color development, and the sections were counterstained with hematoxylin (Solarbio, China), dehydrated, and mounted with neutral gum. All slides were observed and photographed under a microscope (200 or 400×) by a blinded investigator.
Extraction and culture of primary HESCs
Primary HESCs were extracted by 0.2% type I collagenase digestion and filtering with a sieve. Ophthalmic scissors were used to mince the endometrial tissues obtained by curettage. A total of 4-5 ml of 0.2% type I collagenase was added, and the tissue was digested in a 37 °C constant-temperature water bath for 60 min. The solution was filtered through a 200- to 400-mesh sieve, and the suspension was collected. After centrifugation of the suspension at 1000 rpm for 5 min, the supernatant was discarded, and the cells were resuspended in complete medium and placed in a 37 °C, 5% CO2 incubator. After 6-8 h, the medium was replaced (nonadherent cells were also removed), and the purified endometrial stromal cells were obtained. The medium was replaced every 2-3 d, and the cells were passaged at a 1:3-1:4 ratio and cryopreserved [41].
ICC and IF
Cells were fixed in 4% paraformaldehyde for 4 min, washed with PBS three times, incubated with 0.5% Triton-100 at room temperature for 20 min and washed with PBS three times. The cells were blocked in bovine serum albumin (BSA; 5%) for 20 min at room temperature, and then the blocking solution was removed. Antibodies against vimentin and CK-18 were added; instead of a primary antibody, PBS was added to the negative control. The cells were incubated overnight in a humidified incubator at 4 °C. The next day, the primary antibody was discarded, the cells were washed three times with PBS, and a goat anti-rabbit secondary antibody was added. The cells were incubated with the secondary antibody at 37 °C for 20 min, followed by three washes with PBS. Horseradish peroxidase-conjugated avidin (HRP-avidin, SABC) was added, and the cells were incubated at 37 °C for 20 min and then washed with PBS four times. DAB staining was performed in the dark at 37 °C for 5-10 min, followed by termination of the reaction with deionized (DI) water. The nuclei were counterstained with hematoxylin and incubated at 37 °C for 2 min. Staining was terminated with DI water. The samples were dehydrated using a conventional ethanol gradient (75%-85%- 95%-100%) for 2 min at each percentage and vitrified by dimethylbenzene for 1 min. The sections were observed and photographed under an inverted microscope (100×).
Cells were fixed in 4% paraformaldehyde for 15 min, washed with PBS for 5 min (three times), incubated in cell membrane permeabilization solution containing 0.5% Triton X-100 for 10 min, washed with 250 μl of PBS for 5 min (three times) and blocked in 5% BSA for 1 h. The blocking solution was removed, and the cells were incubated in primary antibody in a 4 °C freezer overnight. The next day, the cells were washed with PBS for 5 min (three times) and incubated in the dark for 1-2 h with secondary antibodies conjugated to Alexa FluorTM 488 or Alexa FluorTM 633 (Thermo Fisher, USA). The secondary antibodies were removed, and the cells were washed with 250 μl of PBS for 5 min (three times). The cell nuclei were stained with 4′ 6‐diamidino‐2‐phenylindole (DAPI) for 5 min, and the DAPI was removed. The cells were washed with 250 μl of PBS for 5 min (three times), placed on glass slides (cells facing down) and labeled; glycerin was added to seal the slides. The slides were observed and photographed under an upright or inverted fluorescence microscope using LSCM (Carl Zeiss, LSM 880, Germany) [42] (630× or 1000).
Development of an IUA cell model (TGF-β1-treated HESCs)
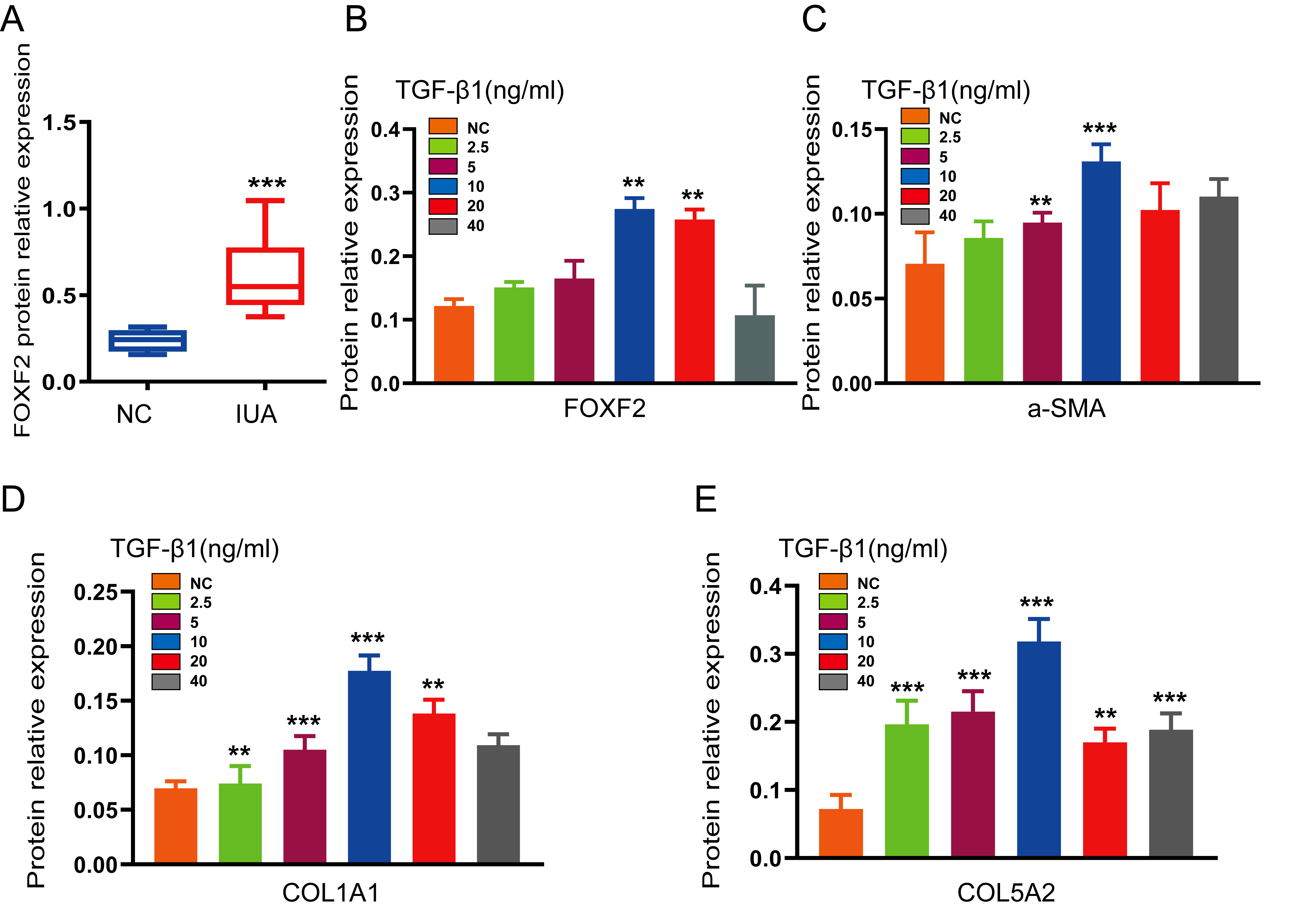
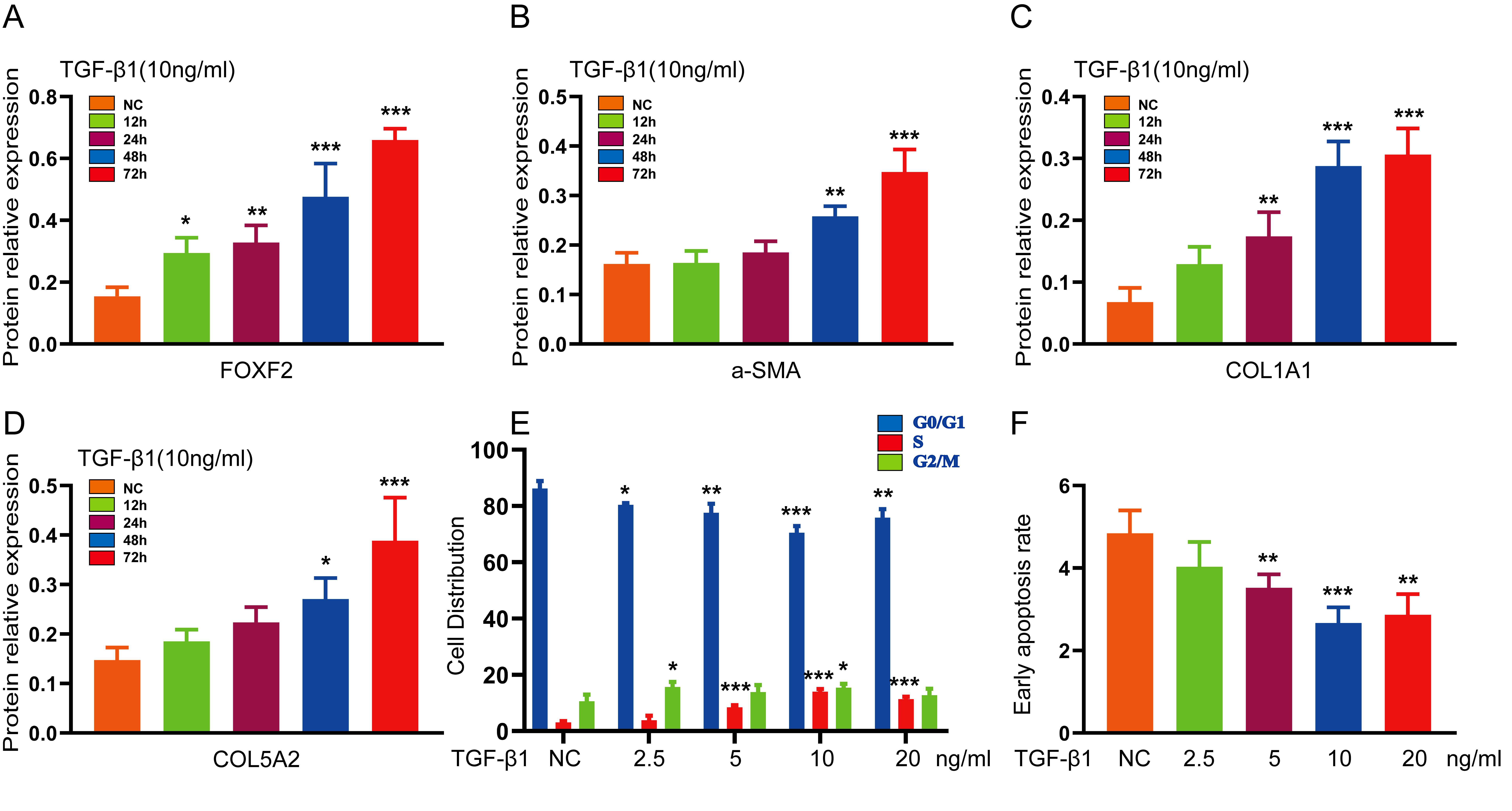
TGF-β1 (0, 2.5, 5, 10, 20 and 40ng/ml) was applied to HESCs for 72 h. The mRNA and protein expression levels of COL1A1, α-SMA, COL5A2 and FOXF2 were detected by qRT-PCR, WB and IF, and cell proliferation and apoptosis were detected by flow cytometry. ECM formation, FOXF2 expression and cell proliferation were most prominent after treatment of the cells with 10ng / ml TGF-β1. HESCs were then treated with 10ng / ml TGF-β1 for 0, 24, 48 and 72 h. The mRNA and protein expression levels of COL1A1, α-SMA, COL5A2 and FOXF2 were detected by qRT-PCR and WB. Treatment of HESCs with 10ng/ml TGF-β1 for 72 h was found to be the optimal condition for development of an IUA cell model.
qRT-PCR
RNA was extracted using TRIzol (Invitrogen), and the purity and concentration of the RNA were determined using a UV spectrophotometer. RNA was reverse- transcribed into cDNA using a PrimeScriptTM RT reagent kit (Takara, Japan). Amplification was performed according to the SYBR® Premix Ex Taq (Takara, Japan) protocol using a CFX96TM Real-Time PCR Detection System (Bio-Rad). Relative quantification was performed using the 2-ΔΔCt method. The 2-ΔΔCt value represents the expression level of a target gene in each group relative to the expression level of the internal reference gene. ΔΔCt = (Ct target gene-Ct reference gene) experimental group-(Ct target gene-Ct reference gene) control group. The Ct value was automatically determined based on the amplification curve. All of the reactions were performed in triplicate. The primer sequences are listed in Supplementary Table 1.
Western blotting (WB)
Total proteins were extracted from primary HESCs or endometrial tissue using RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China). The BCA-100 protein quantitation method (Keygen Biotech, China) was used to determine the protein concentration. An 8-12% separation gel and a 5% stacking gel were prepared for electrophoresis. After separation, the separated proteins were transferred to a membrane by the wet transfer method. The membrane was blocked in 5% skim milk (total protein) and BSA (phosphorylated protein) for 1 h, and primary antibodies (details are shown in supplementary materials) were then added. The membrane was stored at 4 °C overnight and then washed with 1× Tris-buffered saline-Tween (TBST) for 5 min (three times). After addition of the appropriate secondary antibodies, the membrane was incubated at room temperature for 1-2 h and then washed with 1× TBST for 5 min (three times). The membrane was incubated in electrochemiluminescence (ECL) substrate (Millipore, USA), and the blots were developed in an ultrasensitive chemiluminescence imaging system (Bio-Rad) (The antibodies used in these experiments are provided in supplementary Table 2).
Flow cytometry
For cell cycle analysis, cells were trypsinized, resuspended in PBS, and washed twice. The cells were incubated in precooled 70% ethanol, and the ethanol was discarded after centrifugation. The cells were then resuspended in PBS and washed twice. Propidium iodide (PI, 450μl)/RNase (50μl) staining buffer (BD PharmingenTM, USA) was added, and the reaction was allowed to proceed at room temperature for 30 min in the dark. The samples were filtered through a 200-mesh nylon sieve and then sent to a flow detection tube. A FACS flow cytometer was used (Verse, BD, USA).
For apoptosis detection, cells were digested in 0.25% trypsin without ethylenediaminetetraacetic acid (EDTA). The cells were resuspended in PBS and washed twice; 100μl of 1× binding buffer, 5μl of FITC-annexin V (eBioscience, USA) and 10μl of PI (eBioscience, USA) were then added to each sample in the dark. The dye was mixed thoroughly in the dark at room temperature for 15 min and mixed with PBS (400μl / sample). The final volume was 500μl /sample. The samples were filtered through a 200-mesh nylon sieve into a flow detection tube. Each sample was labeled and detected within 1 h of loading.
Chromatin immunoprecipitation (ChIP)
Cells were fixed with 1% formalin to crosslink the protein and DNA. Glycine (Sigma) at a final concentration of 0.125mol/ L was used to terminate the crosslinking reaction. One milliliter of precooled PBS + 1× protein inhibitor cocktail was used to wash the cells. After centrifugation, the supernatant was discarded. Sodium dodecyl sulfate (SDS) lysis buffer (1 ml per 1×107 cells) was then added, and the cells were incubated on ice for 10 min. The chromatin was processed by ultrasonic fragmentation to obtain 200-1500-bp DNA-protein fragments. A total of 5μl of the supernatant was taken as the input group; 45μl of the supernatant was diluted in 450μl of 1× IP dilution buffer, and 500μl of diluted lysate was added to each IP sample for insertion into the plug spin column. The primary antibody (negative control IP: 1-2μl of rabbit anti-human IgG; target-specific IP: 1-10μg of rabbit anti-human polyclonal FOXF2 antibody) was added, and the samples were mixed thoroughly at 4 °C overnight. Then, 20μl of ChIP-grade protein A/G plus agarose was added to the immunoprecipitation reaction, and the mixture was incubated at 4 °C in a shaker for 1 h. The cells were washed and eluted according to the instructions. The IP group was mixed thoroughly with 2μl of RNase A and 5μl of proteinase K. A total of 150μl of 1× IP elution buffer, 2μl of RNase A and 5μl of proteinase K were added to the input group samples. Decrosslinking of the protein-DNA complex was conducted in a metal bath at 65 °C for 3 h. The DNA fragments were purified using a kit for high-throughput sequencing and qRT-PCR.
Cell transfection
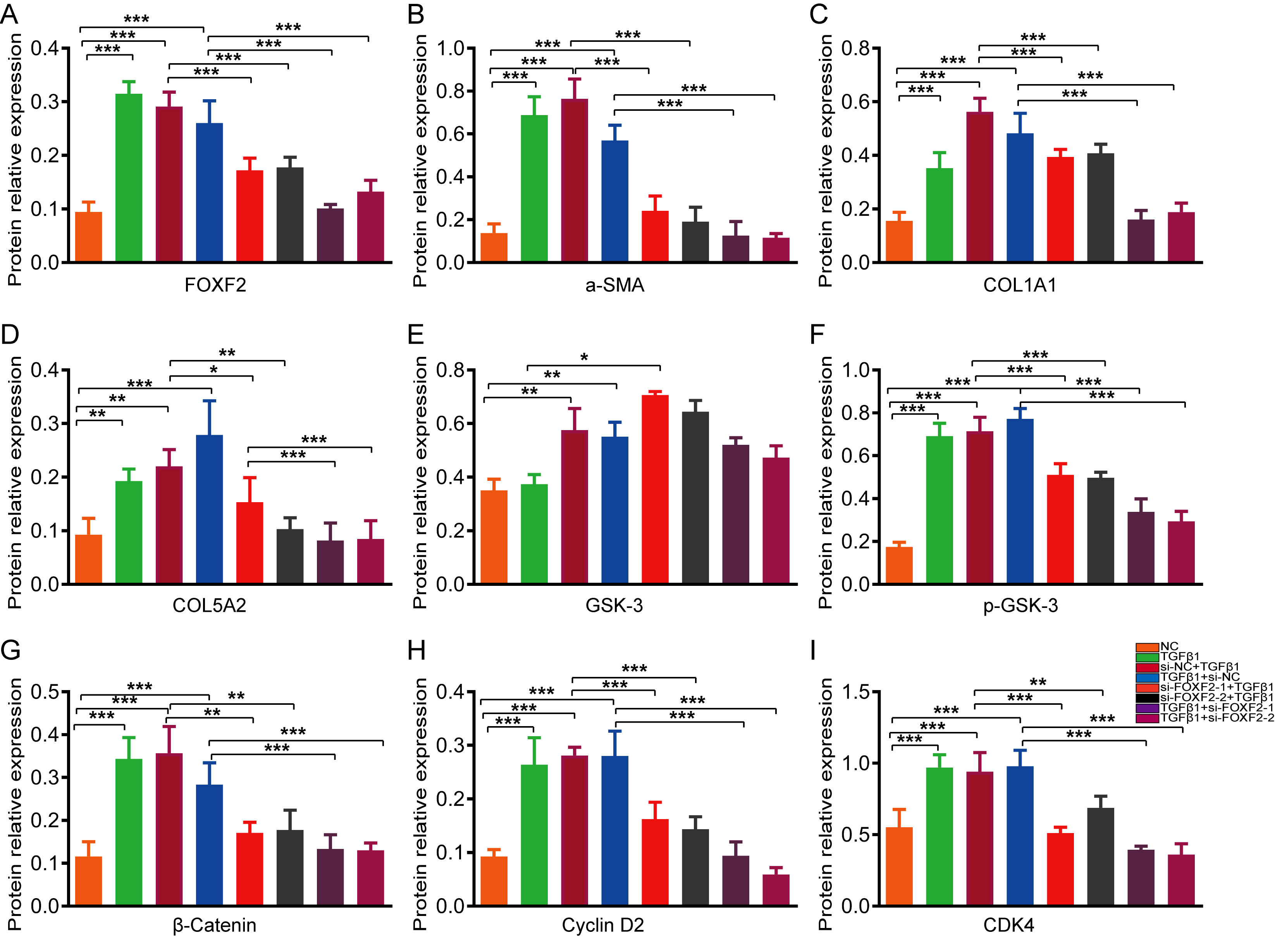
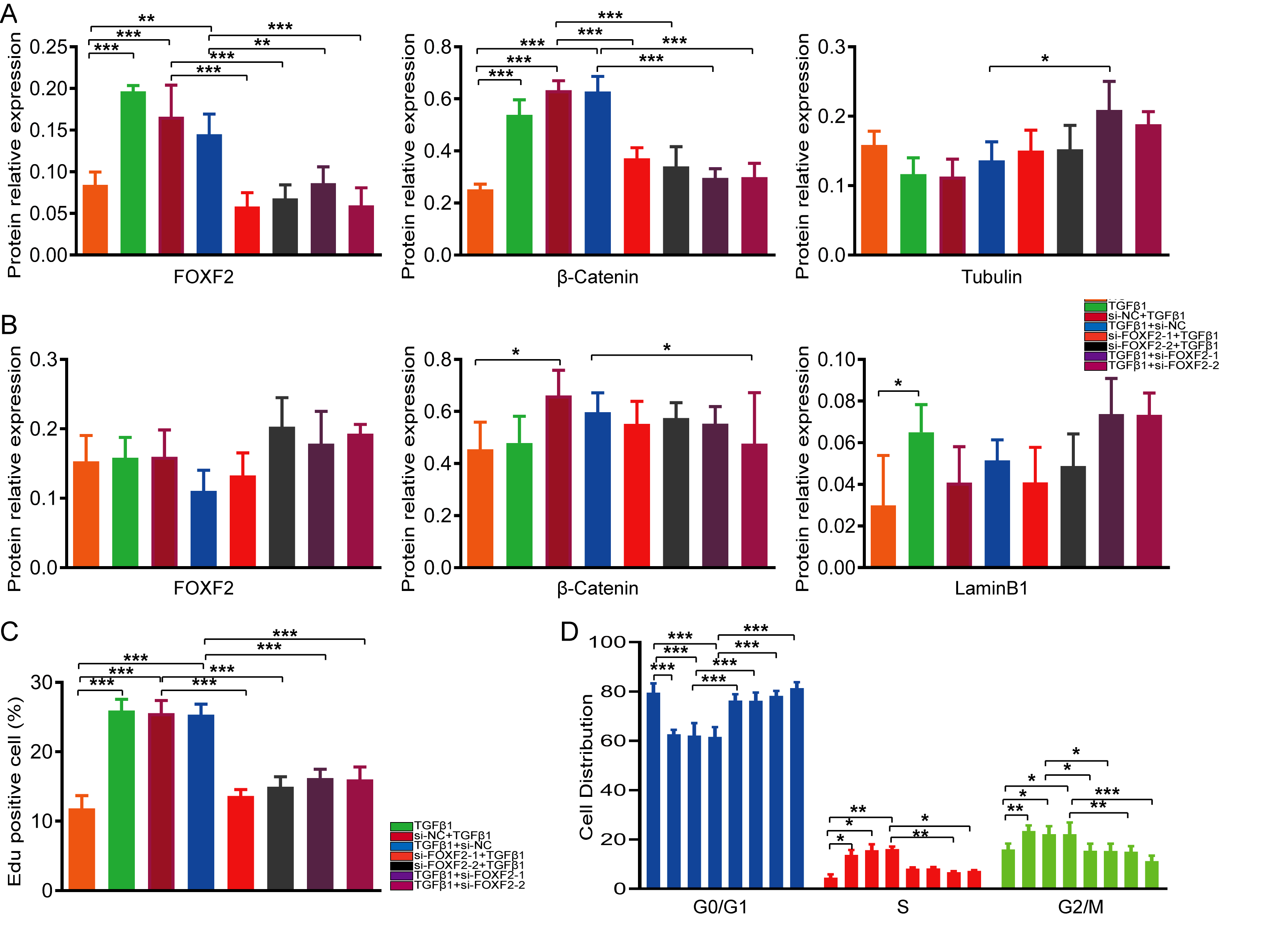
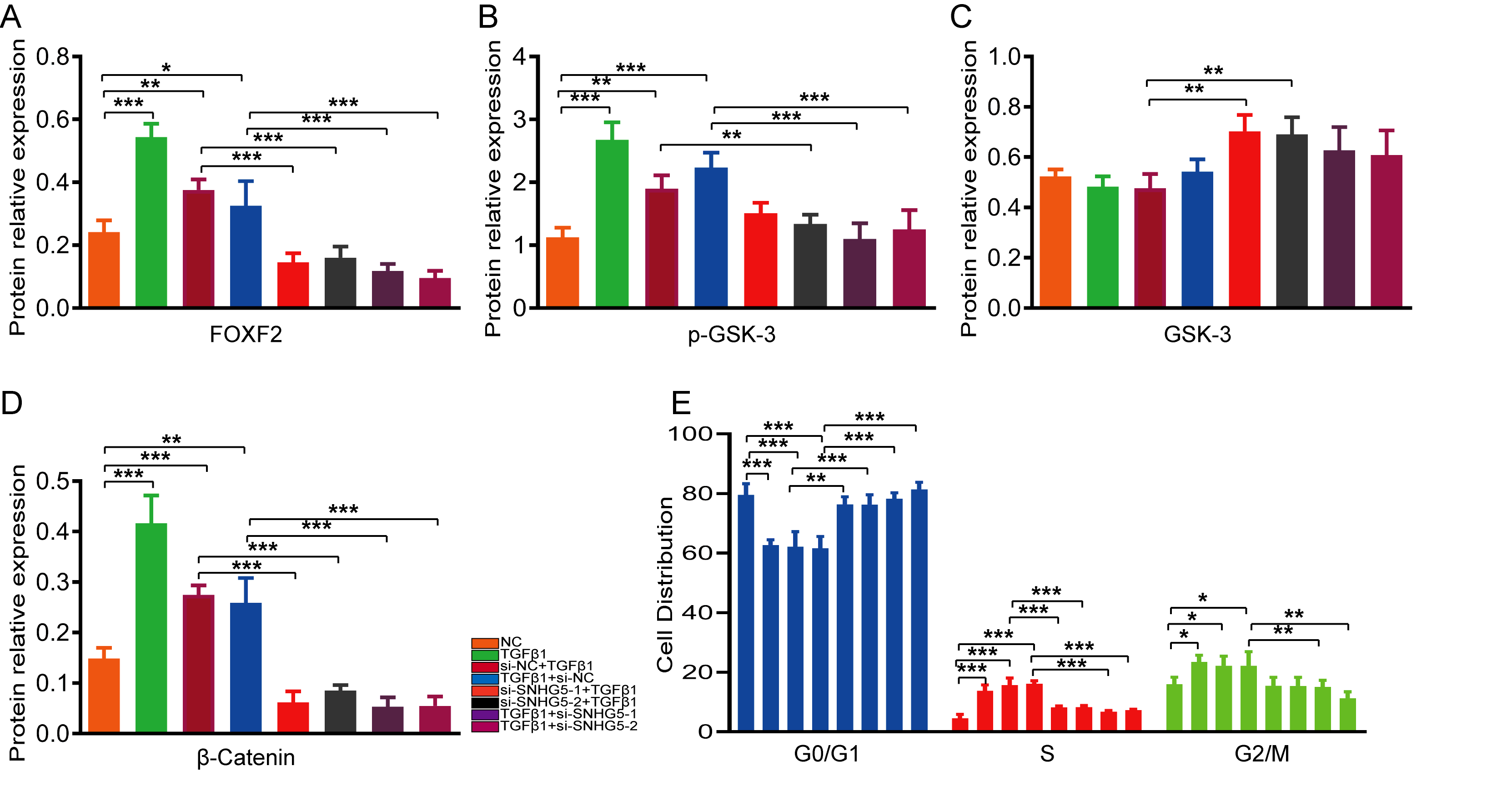
For the prevention group, HESCs were incubated overnight to allow them to reach 20-30% confluence and then cultured in serum-free medium for 24 h to synchronize the cell cycle. siRNA and si-NC designed by GenePharma (Shanghai, China) were transfected into the cells using Lipofectamine® 3000 (Invitrogen; Thermo Fisher Scientific) according to the manufacturer’s instructions. Twelve hours after transfection, the HESCs were stimulated with culture medium containing 10ng / ml TGF-β1 for 72 h.
For the treatment group, after achieving a synchronous cell cycle, HESCs were treated with 10 ng /ml TGF-β1 for 48 h. The HESCs were then transfected with siRNA or si-NC for 12 h and maintained in medium containing 10ng /ml TGF-β1 for 36 h.
EdU and CCK8 assays
EdU assay: Cells were incubated for 4 h in 100 μl of complete medium (without antibody) to which 0.2μl of EdU working solution (CWBiotech) had been added. They were then fixed in 4% paraformaldehyde for 15 min, in 50μl of glycine for 5 min, and washed twice with 3% BSA. To permeabilize the cell membrane, the cells were incubated in 0.5% Triton X-100 for 20 min and washed twice with 3% BSA. According to the instructions supplied by the manufacturer, the prepared mixture was added for 30 min, and the cells were washed twice with 3% BSA and once with PBS. The final concentration of Hoechst 33342 was 5μg /ml. The cells were incubated in the dark for 15 min and washed twice with PBS. Images were captured using an inverted fluorescence microscope in a darkroom (100×).
CCK8 assay: Six hours after the cells were seeded in a 96-well plate, the supernatant was removed, only the cells that adhered tightly to the wells. CCK8 (10μl) and basal medium (90μl) were added to each well to yield a total volume of 100μl. A blank control plate containing no cells but the same volumes of CCK8 and basal medium was also prepared. The cells and control plates were incubated for 2-3 h, and the optical density (OD) of each well at a wavelength of 450 nm was measured in the dark every 30 min. A growth curve was plotted according to the measured OD values.
RIP and RNA pulldown assays
RIP: The RIP experiment was performed using a RIPTM RNA-binding protein immunoprecipitation kit (Millipore, USA) according to the manufacturer's instructions. Approximately 5μg of antibody (target protein, FOXF2; negative control protein, rabbit IgG; positive control protein, SNRNP70) was added, and the sample was incubated with protein G magnetic beads. After the addition of cell lysis buffer, the coprecipitated RNA was pulled down with protein G beads, followed by high-throughput sequencing.
RNA pulldown: Biotin-labeled SNHG5 was synthesized using T7 RNA polymerase in a biotin RNA-labeled mixture (Roche, USA) and incubated with cell lysate for 4 h. After overnight incubation with streptavidin-coated magnetic beads (Thermo, USA), biotin-labeled SNHG5 protein was pulled down. Specific bands were identified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), silver staining, and WB. The primer sequences are listed in Table S1 in the supplementary materials.
FISH
Cells on slides were fixed in 4% paraformaldehyde for 10 min and washed with PBS for 5 min (three times). The cells were incubated in precooled cell membrane permeabilization solution (0.5% Triton X-100) at 4 °C for 5 min and then washed with PBS for 5 min (three times). A total of 200μl of prehybridization solution was added, and the cells were incubated at 37 °C for 30 min. All subsequent operations were conducted in the dark. A total of 2.5μl of 20μM lncRNA SNHG5 FISH Probe Mix stock solution was added to 100μl of hybridization solution, and the cells were incubated with the solution in a 37 °C incubator overnight. The slides were then washed with hybridization solution I for 5 min at 42 °C (three times), once with hybridization solution II at 42 °C, once with hybridization solution III and once with PBS. After DAPI staining for 10 min, the slides were washed with 500μl of PBS for 5 min (three times). The slides were removed, mounted on glass in enhanced fluorescence signal substrate, and photographed under inverted LSCM (630×).
Separation of the nucleus and cytoplasm
The cells were washed twice with precooled PBS, resuspended in cell fractionation buffer, and incubated on ice for 5-10 min or until a clear solution was obtained. The cells were centrifuged at 500×g for 5 min at 4 °C to precipitate the nuclei; the cytoplasm remained in the top fraction. An equal volume of 2× Lysis/Binding Solution was added to the cytoplasm. An equal volume of anhydrous ethanol was added. The sample was filtered, and the filtrate was discarded. The sample was washed once with Wash Solution 1 and twice with Wash Solution 2/3. Elution Solution that had been heated to 95 °C was then added. The sample was centrifuged for 30 s to obtain the RNA. Elution Solution was added again, and the sample was centrifuged for 30 s. The nuclear RNA and cytoplasmic RNA were stored at -80 °C or used for qRT-PCR.
PPI network construction
The PPI information was predicted using the Search Tool for the Retrieval of Interacting Genes (STRING) online database (http://string-db.org). Analyzing the functional interactions among proteins could offer insights into the mechanisms of occurrence or development of IUA. To evaluate the potential PPI interaction, PPI network of DEGs was mapped using STRING database, and the relationship with a combined score > 0.4 was considered statistically significant.
Statistical analysis
Statistical analysis was performed using SPSS 20.0 (Chicago, USA) statistical software. One-way analysis of variance (ANOVA) was used to evaluate differences between two or multiple groups. Measurement data are expressed as the mean ± standard error of the mean (SEM) obtained in one representative experiment out of three independent experiments. P < 0.05 was considered statistically significant. Figures were generated with GraphPad Prism 7 (GraphPad Software, USA) and Adobe illustrator CS6 (Adobe, USA).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}