Human stromal vascular fraction (hSVF) isolation from human adipose tissue and purification of human pericytes from hSVF
Human adipose tissue was obtained from eight patients who underwent total knee arthroplasty due to degenerative osteoarthritis (Supplemental Table A). The adipose tissue samples were stored (4°C) until they were processed. All samples were processed within 48 h after collection. Human stromal vascular fraction (hSVF) was prepared by digesting the adipose tissue using collagenase digestion, as previously described [19]. The adipose tissue was briefly washed in an equal volume of phosphate buffered saline (PBS). Collagenase digestion was performed using Dulbecco's Modified Eagle's Medium (DMEM; Sigma-Aldrich, St. Louis, MO, USA) containing 3.5% bovine serum albumin (Sigma-Aldrich) and 1 mg/ml collagenase type II for 70 min under agitation at 37°C. The filtered solution was centrifuged to separate and exclude adipocytes. The processed hSVF was suspended in red cell lysis buffer (155 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA) and incubated for 10 min at room temperature. The hSVF filtrate was immediately processed for human pericyte purification.
A fluorescence-activated cell sorter (FACS) was used to purify human pericytes from isolated hSVF, as previously described [20, 21]. The isolated hSVF was centrifuged and the resulting pellet was incubated (4°C for 15 min in the dark) with conjugated antibodies (anti-CD34-phycoerythrin (Dako), anti-CD45-allophycocyanin (Santa Cruz Biotechnology, Inc., Dallas, TX, USA), and anti-CD146-fluorescein isothiocyanate (AbD Serotec)). The hSVF pellet was then resuspended in PBS and 4', 6-diamidino-2-phenylindole (Invitrogen, Carlsbad, CA, USA) and was filtered through a 70 µm cell filter for removal of nonviable cells. The solution was processed on a FACS Aria cell sorter (BD Biosciences, San Jose, CA, USA) to isolate populations of cells that constituted human pluripotent stem cells, based on cell surface markers: pericytes (CD146+, CD34-, CD45-).
Osteogenic and adipogenic differentiation of human pericytes
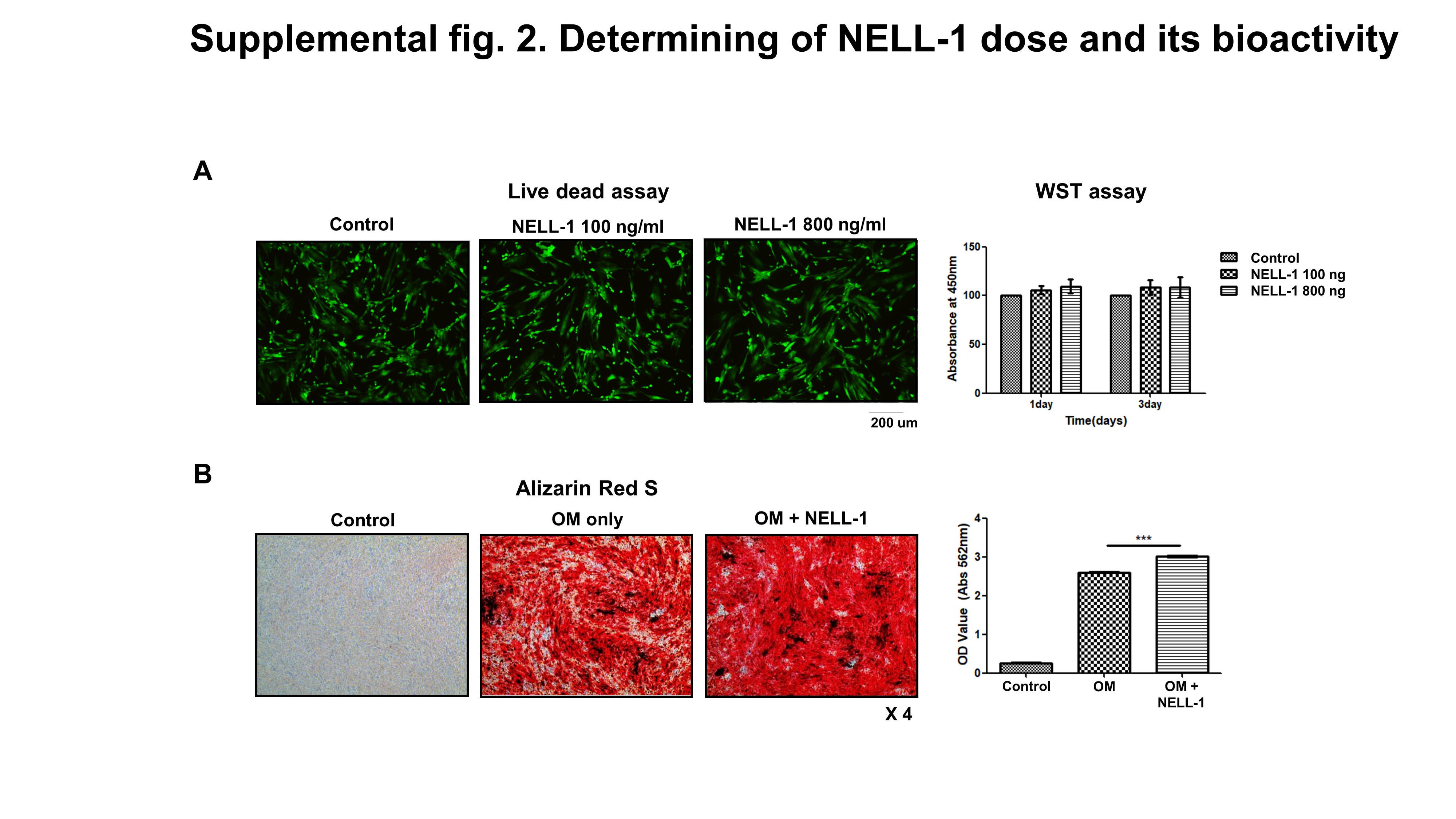
The human pericytes were cultured separately in the PBS control and NELL-1 solutions to determine the differentiation potential of human pericytes and to test the biologic activity of NELL-1 protein during osteogenesis and adipogenesis. Osteogenic differentiation of the pericytes occurred over a period of 15 d. The cells were added to 24-well plates (3 × 104 cells/well density) with DMEM + 10% fetal bovine serum (FBS). Within 24 h, osteogenic differentiation of the cells was induced in the PBS control and NELL-1 (800 ng/ml) treatments using osteogenic differentiation medium (DMEM + 10% FBS + 50 µg/ml ascorbic acid, and 3 mM β-glycerophosphate). The medium was changed every 3 d. Alizarin Red staining was used to assess osteogenic differentiation.
For adipogenic differentiation, human pericytes were added to 24-well plates (5 × 104 cells/well density) with DMEM + 10% FBS. In 24 h, cells were induced to adipogenic differentiation in the PBS control and NELL-1 (800 ng/ml) treatments using adipogenic differentiation medium (Human MesenCult™ Adipogenic Differentiation Medium; STEMCELL TECHNOLOGIES, Catalog #05412). Adipogenic differentiation was performed over 12 d. The medium was changed every 3 d. Adipogenic differentiation was assessed at 12 d using Oil Red O staining.
Endothelial cell culture
Human umbilical vein endothelial cells (HUVECs) were purchased from Lonza and cultured in endothelial growth medium (Lonza, Basel, Switzerland). The cells were used between passages 4 and 7 [22].
Viability test
To determine the dose of NELL-1 protein, we performed a viability test after adding various doses of NELL-1. Two thousand pericytes were added to 100 µl DMEM in each well of two 96-well plates. The cells were incubated in DMEM with 100 ng/ml or 800 ng/ml NELL-1 for 72 h. Control cells were incubated with only DMEM. A water-soluble tetrazolium salt (WST) assay (Cell Counting Kit-8; Dojindo, Kumamoto, Japan) was used to measure cell proliferation. Then, WST (10 µl) was added to each well and the cultures were incubated for an additional 2 h at 5% CO2 and 37°C before evaluation using spectrophotometry. Conversion of WST to formazan was measured at 450 nm. The results were normalized and were presented as percentage of the viable cells in the control group [23, 24].
Tube formation assay
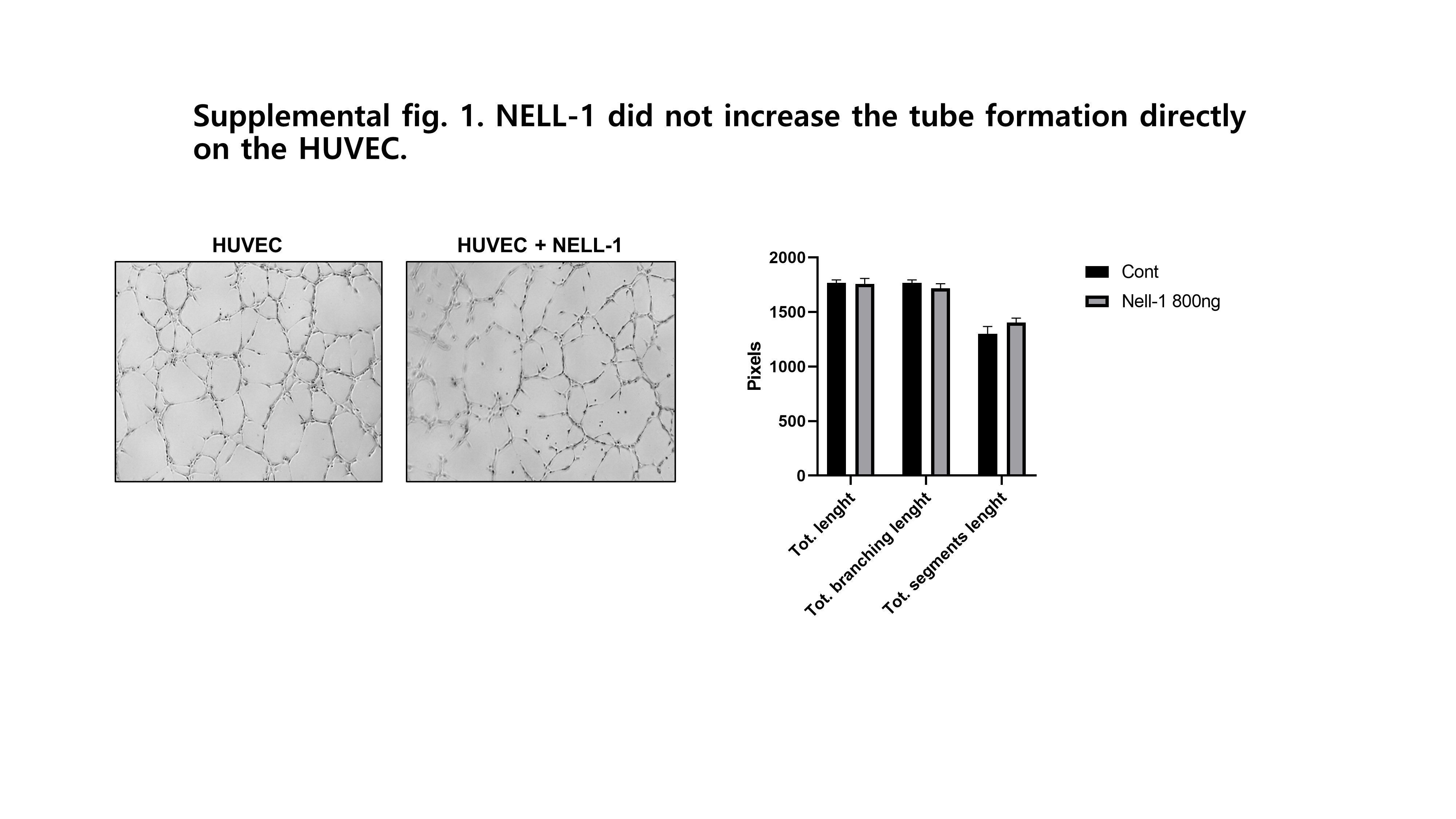
A capillary tube formation assay (Matrigel, Corning, Corning, NY, USA) was used to assess the effects of human pericytes and NELL-1 on endothelial cell morphogenesis. Briefly, 4 × 104 cells per well were added to Matrigel-pre-coated 96-well plates and treated with pericytes (0.2 × 104 cells/well) or NELL-1 (800 ng/ml), or both. Suramin (40 mM; Sigma-Aldrich) was included as a negative control. After 12 h, cell changes were recorded using a microscope (Nikon ECLIPSE Ti2) and analyzed using the Image J software program. This program provided automated quantitative measurements of tube characteristics (e.g., number of connected tubes, tube area, and angiogenic index).
Wound migration assay
HUVECs were added at a density of 3 × 103 cells per well in the ibidi Culture-Insert 2 Well in a µ-Plate 24 Well plate and allowed to grow into a confluent monolayer overnight. Fresh medium containing indicated concentrations of human pericytes and NELL-1 was added. The Culture-Insert 2 Well was then removed with sterile tweezers. After 24 h, the cells were photographed using a JuLI™ Stage Cell History Recorder (NanoEntek, Guro-gu, Seoul, Korea). Migration rate was calculated using the JuLI™ Stage. Three measurements of three independent wounds were taken for each monolayer sample.
Chick chorioallantoic membrane assay
The chick chorioallantoic membrane (CAM) assay was used to assess the effect of NELL-1 on ex vivo angiogenesis. Briefly, fertile chicken eggs were candled on embryonic day 3. A small opening was made at the top of the live eggs and a sterilized Thermanox™ Coverslip (Nunc™) saturated with either PBS or NELL-1 was placed on the CAM. The holes were then sealed with cellophane tape. The eggs were photographed after a 72-h incubation. Image J software was used to quantify blood vessel density; the results were presented using bar diagrams [24].
Animals
Twenty-four 8-week-old male NOD SCID mice (CHA Institute Animal Experimentation, Pangyo, Seongnam, Korea) were used for the study to prevent immune reactions to implants containing human cells. Each mouse was housed alone in a pathogen-free ventilated cage, fed a standard rodent chow diet, provided tap water ad libitum, and experienced 12-h light and dark cycles. The mice were cared for following the Chancellor’s Animal Research Committee for Protection of Research Subjects guidelines at the CHA medical university (IACUC190082).
Implant preparation and grouping
Recombinant human NELL-1 was purchased from Bone Biologics Inc. (UCLA, CA, USA). An absorbable collagen sponge (ACS) of defined dimensions (0.5 cm × 0.5 cm × 1.0 cm, Lyoplant, 1066102; Aesculap AG, Tuttlingen, Germany) was used. Because intramuscular implantation of ACS alone has no known bone-forming effects, this carrier was chosen for its nonosteoinductive characteristics [25]. Defined concentrations of viable cells and NELL-1 in PBS suspension (20 ul) from the hSVF were applied and allowed to saturate the ACS. The cell and protein scaffold suspensions were kept on ice until implantation. The four treatment groups used were (a) control with PBS (b) high concentration (800 ng/ml) NELL-1, (c) Pericytes, and (d) Pericytes loaded with a high concentration (800 ng/ml) of NELL-1.
Surgical procedure for mouse models of osteonecrosis
We used a mouse model with a necrotic bone fragment to replace osteonecrosis in humans. A total of 30 mice (6 for control, 8 for NELL-group, 8 for Pericytes group, and 8 for Pericytes + NELL-1 group) were used for the experiment. All mice were prepared at the age of 3 months. Anesthesia was initiated in a 5% gaseous isoflourane-filled holding chamber and maintained with 3–4% gaseous isoflourane through a nose cone. Before making an incision, the thigh of the mouse was shaved, and the skin prepared using an alcohol and betadine solution. Sterile ophthalmic lubricant ointment was applied to each eye and a buprenorphine injection (0.05 mg/kg) was given via the subcutaneous route.
All surgical procedures were performed by the senior author, an experienced orthopedic surgeon. The femoral bone was exposed using an incision made on the anterolateral aspect of the thigh. A PEEK plate was located on the anterior femur. The most proximal hole of the plate was gently drilled using a 0.3 mm drill bit and the first screw was inserted. Additional distal screws were inserted in a similar fashion. The 0.22 mm Gigli saw wire was closely placed around the bone in a medio-lateral orientation and inserted in the slots of the customized jig to create a 2.5-mm bone defect. The jig was inserted on the stem of the two last screws and applied above the plate. Next, a 2.5-mm long mid-diaphyseal femoral ostectomy was performed using the Gigli saw while applying a constant steady tension. Care was taken to avoid excess movement to obtain a straight bone cut. After the ostectomy, the Gigli saw was removed and the saw wire was cut close to the bone on one side. The jig and remaining stems of the screw were removed. The bone fragment from the ostectomy was removed and immersed in liquid nitrogen for 5 min to induce necrosis. The ACS treated with the assigned material based on treatment group (± Pericytes ± NELL-1) was put below the femoral shaft and the necrotic bone fragment was taken back to the original site. It was then wrapped using the ACS. Finally, the ACS was sutured to prevent displacement of the necrotic bone fragment and the wound was closed (Fig. 1).
In vivo plain radiograph
In vivo bone regeneration was assessed using plain radiography images (GIX-I, Genoray, Seongnam, Korea; Ultra Light Portable X-ray, Nanoray, Deagu, Korea) of the femur under the conditions of 70 kV/20 mA, 0.06 sec; 20 lines per mm spatial resolution. Standard lateral digital radiographs of the femur were taken immediately after surgery and 4 weeks later under volatile anesthesia [26].
Ex vivo micro computed tomography
The mice were euthanized using CO2 asphyxiation 4 weeks after surgery. High-resolution micro computed tomography (CT) scanning of each sample was performed (Bruker microCT Skyscan 1173). Each piece of femur bone was in a polyethylene tube filled with alcohol (75 volume percent) during scanning. The radiographic projections were acquired at 130 kV and 60 uA with a fixed exposure time of 500 ms, an A1 1.0 mm filter, and a 6.04 um pixel size. Four frames were averaged for each rotation increment of 0.9. Three-dimensional images with a voxel average size of 13 mm were reconstructed using a Hamming-filtered back-projection, and the manufacturer’s reconstruction software (NRecon; Skyscan, Aartselaar, Belgium). Bone mineral density (BMD) of the femur was measured using the Bruker microCT with a phantom. The analyses were performed in the same manner for each mouse, with a volume of interest corresponding to the respective defect. The number of united cortices in two orthogonal reconstructed views was recorded for the qualitative analysis. Bone union was defined as union of four out of four cortices. Resident software (CTAn; Skyscan, Aartselaar, Belgium) was used to obtain the BV/TV and BMD for quantitative analysis of bone formation within a region of interest. A lower gray threshold of 45 grayscale indices (attenuation coefficient of 0.035) and an upper gray threshold of 240 grayscale indices (attenuation coefficient of 0.186) were used for each mouse [27].
Histology and histomorphometric analysis
The animals were euthanized 4 weeks after surgery. Histologic specimens were fixed in 4% paraformaldehyde at 4°C for 1 d, followed by decalcification (Calci-Clear™ Rapid Decalcifying Solution, HS-105; National Diagnostics) for 3 h at room temperature with gentle mechanical stirring. The specimens were then dehydrated and embedded in paraffin. The tissue blocks were sectioned to 3-mm thicknesses along longitudinal planes (Leica RM2235 microtome; Leica Microsystems GmbH, Wetzlar, Germany). All sections were stained with hematoxylin and eosin and alcian blue stain.
Microarray
Microarrays were used to measure expression levels of genes related to NELL-1 activity. NELL-1 treated Pericyte samples were used. Briefly, total RNA was isolated using RNeasy columns (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocol. The RNA samples were quantified after processing with DNase digestion and clean-up procedures. An Ambion Illumina RNA amplification kit (Ambion, Austin, TX, USA) was used to amplify and purify the total RNA. Total RNA (550 ng) from each sample was converted to double-strand cDNA. Using a T7 oligo (dT) primer, amplified RNA (cRNA) was generated from the double-stranded cDNA template using an in vitro transcription reaction and purified using the Affymetrix sample clean-up module. An ND-1000 Spectrophotometer (NanoDrop, Wilmington, DE, USA) was used to quantify cDNA after purification. Uracil-DNA glycosylase and apurinic/apyrimidinic endonuclease and restriction endonucleases were used to fragment the cDNA. It was end-labeled using a terminal transferase reaction incorporating a biotinylated dideoxynucleotide. Fragmented end-labeled cDNA was hybridized to the GeneChip Human Gene 2.0 ST arrays manual (Affymetrix, Santa Clara, CA, USA). After hybridization, the chips were stained and washed (GeneChip Fluidics Station 450; Affymetrix) and scanned (GeneChip Array scanner 3000 G7; Affymetrix) by Macrogen Ltd. (Seoul, South Korea). Affymetrix® GeneChip™ Command Console software was used to compute signal values.
RNA isolation and qRT-PCR analysis
Total RNA was extracted from cells using Trizol reagent (Invitrogen), according to the manufacturers' instruction. One micrograms of total RNA were used to determine the expression of mRNAs using AMPIGENE® qPCR Green Mix (Enzo Biochem, Inc.) and iCycler real-time PCR detection system (Bio-Rad, CA) according to the manufacturers' instruction. The sequences of the primers were as follows: FGF2, 5′-AGAAGAGCGACCCTCACATCA-3′ (forward) and 5′-CGGTTAGCACACACTCCTTTG-3′ (reverse); IL-6, 5′-ACTCACCTCTTCAGAACGAATTG-3′ (forward) and 5′-CCATCTTTGGAAGGTTCAGGTTG-3′ (reverse); TGFB2, 5′- CAGCACACTCGATATGGACCA-3′ (forward) and 5′-CCTCGGGCTCAGGATAGTCT-3′ (reverse); VEGFA, 5′-AGGGCAGAATCATCACGAAGT-3′ (forward) and 5′-AGGGTCTCGATTGGATGGCA-3′ (reverse); β-actin, 5′-ACCGAGCGCGGCTACAG-3′ (forward) and 5′-CTTAATGTCACGCACGATTTCC-3′. β-actin was used for normalization of mRNA.
Western blot
Protein extraction buffer (Pro-Prep, iNtRON Biotechnology, Gyeonggi-do, Korea) was used to lyse cells for western blot. After centrifugation (4°C, 13,000 rpm for 15 min), protein content of lysed cells was assessed (Bradford assay). Equal total protein amounts were run on 12% SDS polyacrylamide gels and transferred to a nitrocellulose membrane. Membranes were blocked using 5% non-fat milk powder at room temperature and then incubated with primary antibodies in tris-buffered saline-tween 20 (TBS-T) overnight at 4°C. The primary antibodies FGF2 protein 1:1000 (Santa Cruz Biotechnology) were used in the assay. The membranes were then washed in TBS-T and incubated with 1:5000 goat anti-mouse IgG (Santa Cruz Biotechnology) secondary antibodies for 1 h at room temperature. An enhanced luminol-based chemiluminescence detection kit (Bio-Rad Laboratories, Hercules, CA, USA) was used to visualize the resulting bands. Protein quantification was performed using densitometric digital analysis of the protein bands (ChemiDoc™ XRS + with Image Lab™ Software ver. 6.0; Bio-Rad Laboratories). The membrane was re-probed with GAPDH (Santa Cruz Biotechnology) to confirm equal loading. After each sample was run at least three times on western blot analysis, densitometry analysis was performed for each of the three bands.
Statistical analysis
All numerical results were expressed as mean ± standard deviation (SD) values. Analysis of variance was used for comparisons between treatment groups; LSD was used as a post-hoc test or Student’s t-tests were performed. The statistical software IBM SPSS ver. 23.0 (IBM Corp., USA) was used for the analysis, and the significance level was set at P < 0.05.
{kind=link}
{kind=link}