Chemicals
Histamine dihydrochloride, putrescine dihydrochloride, cadaverine dihydrochloride, peroxidase, and N-Ethyl-N-(2-hydroxy-3-sulfopropyl)-3,5-dimethoxyaniline (DAOS) were obtained from Wako Pure Chemical (Japan). Spermidine trihydrochloride, and spermine tetrahydrochloride were purchased from Sigma Chemicals (USA). 4-aminoantipyrine was purchased from Biobasic Chemical Industries (Canada). Betahistine supplied from Shahredaru pharmaceutical co. (Iran). All reagents were analytical grade.
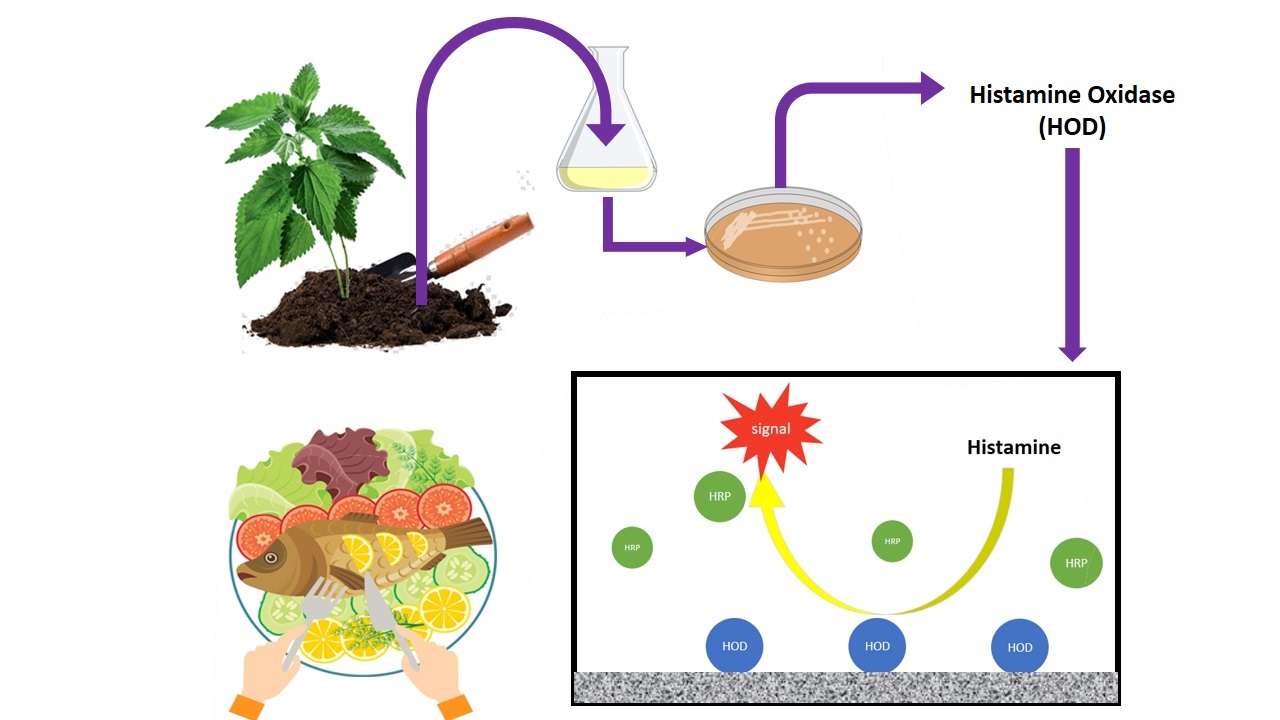
Isolation of histamine metabolizing bacteria
To isolate the histamine-degrading bacteria, different soil samples around the roots of stinging nettle (Urtica dioica) were collected from different regions of Iran. A spoonful (approximately 5 g) of each soil sample was suspended in 100 ml of a screening medium consisting of 0.1 % (w/v) histamine dihydrochloride, 0.05% (w/v) K2HPO4, 0.02% (w/v) MgSO4·7H2O, 0.001%(w/v) FeSO4·7H2O in tap water at pH 7.0. After incubation at 30 °C for seven days, one mL of culture was inoculated in 100 mL of the same fresh medium and incubated for the next two days in the same condition. This step was repeated two times. After enrichment, 0.1 ml of the culture medium was plated out onto agar-solidified screening medium and incubated at 30 °C. The colonies appeared on the plates after 24-48 h, were transferred to 20 ml of Luria-Bertani (LB) medium (tryptone 10 g/L, yeast extract 5 g/L, NaCl 10 g/L), and cultivated at 30 °C, 115 rpm for 12 hours.
Amplification of putative HOD gene and extraction of HOD enzyme
The DNA sequence of HOD from the different bacterial genus (Supporting information Table S1) was obtained from NCBI GenBank and aligned by ClustalW online software (http://www.ebi.ac.uk/Tools/msa/clustalo). The identified conserved sequences were used to design the PCR primers for the amplification of newly isolated HOD genes. PCR amplifications were performed by applying the initial denaturation at 95 °C for 5 minutes, followed by 30 cycles of amplification (94 °C for 40 s, 52 °C for 40 s, and 72 °C for 60 s), and with 7 min of final extension at 72 °C. Genomic DNA of isolated histamine utilizing bacteria was used as template DNA and amplified using the designed primers, (forward: 5ʹ- AACTACGAYTACGGSTTCTACTGG-3ʹ and reverse: 5ʹ- GCATGATSGGCCAGTCCTC-3ʹ). After electrophoresis of PCR results, the bands representing the putative HOD gene were purified from agarose gel using QIAquick purification kit (Qiagen, Italy) and sent for sequencing at Bioneer Co. (Korea).
The resulting sequences were blast-analyzed to detect the sequence similarities. Besides, the PCR positive strains were cultivated in 10 ml of the screening medium for 12 h. The grown cells were harvested by centrifugation at 4,000 g for 10 min at 4 °C and washed twice with 0.1 M phosphate buffer pH 7.0. The resulting cells were resuspended in the wash buffer and sonicated for a total of 5 min (2 s sonications followed by a 10 s intervals) at a power setting of 20 kHz under ice-cooling and using a bench-top sonicator. The cells and debris were removed by centrifugation at 12,000 g for 10 min at 4 °C, and supernatants were collected and referred to as the crude enzyme extract.
Determination of HOD activity
The HOD assay was performed according to Sekiguchi et al. (2004) with minor modifications. The assay mixture (170 μL) consisting of 1.2 mM histamine dihydrochloride, 1.47 mM DAOS, 2.2 mM 4–AA and one unit/mL horseradish peroxidase in 20 mM potassium phosphate buffer (pH 7.0) was preincubated at 37 °C for 5 min. The obtained cell lysate was diluted with 20 mM phosphate buffer (pH 7.0), and 30 μL of the sample was mixed in the assay mixture. The mixtures were incubated for 5 min at 37 ˚C, and the OD600 was measured using an Infinite M200 microtiter plate reader (Switzerland). The mixture without substrate was considered as negative control. One unit of activity was defined as the amount of enzyme which liberated 1µmol of hydrogen peroxide per min under the specified conditions. The amount of H2O2 was calculated using the standard curve.
Taxonomic identification of HOD producing bacterium
The isolated strain with the highest HOD activity was identified by 16S rRNA sequencing. The genomic DNA was amplified by PCR using the following primers of 16S rRNA; 27 F (5′-AGAGTTTGATCMTGGCTCAG-3′), and 1492 R (5′-GGYTACCTTGTTACGACTT-3′). The PCR program comprised initial denaturation at 95 °C for 5 minutes, followed by 30 cycles of amplification (94 °C for 40 s, 52 °C for 40 s, 72 °C for 60 s), and 7 min of final extension at 72 °C. PCR product was purified by the QIAquick purification kit (Quiagen, Italy) and sequenced by Bioneer Co. (Korea). The obtained sequence was compared with reference 16S rRNA gene sequences available in EzBioCloud database (https://www.ezbiocloud.net/). All the representative sequences were aligned using the Clustalx software package, and the phylogenetic tree was constructed using neighbor-joining method available in MEGA version 10 software. Bootstrap analysis based on 1000 replications was conducted for evaluating the confidence level of the branch nodes.
Growth profile of selected bacterium and HOD production
The isolated bacterium was cultivated in LB medium at 30 °C and 115 rpm for 12 h. The grown cells were harvested by centrifugation at 10000 g for two min, washed twice with saline and diluted to match the 0.5 McFarland turbidity standard (OD600=0.13, approximately 108 CFU/ml). Ten ml of the prepared dilution was used for inoculation of one L of basal salt medium (BSM) (0.05% K2HPO4, 0.02% MgSO4.6H2O, 0.001% FeSO4.7H2O, 0.001% CaCl2, and 0.1% (v/v) trace elements (70 mg ZnCl2, 100mg MnCl2.4H2O, 200mg CoCl2.6H2O, 100mg NiCl2.6H2O, 20mg CuCl2.2H2O, 50mg, 50mg NaMoO4.2H2O, 26mg Na2SeO3.5H2O and one ml of 25% HCl in 1000 ml distilled water, pH 8.0)) containing 0.1% histamine dihydrochloride and 1% sodium acetate, and incubated for 18 h at 30 °C, and 115 rpm. Ten mL of cultures were aliquot at regular intervals for monitoring the cell growth by measuring the optical density at 600 nm. Cell lysates were used for the measurement of HOD activity.
Substrate specificity analysis
Substrate specificity of the crude enzyme was checked by using different BAs, including histamine, tyramine, putrescine, cadaverine, spermine, and spermidine, with the final concentration of 1.2 mM. Enzymatic activity on each substrate was determined based on the method described earlier.
Effect of temperature and pH on enzyme activity
The effect of temperature on isolated HOD was examined by incubating 30 µL of the diluted enzyme with 170 µL substrate solution (1.2 mM histamine dihydrochloride in 20 mM potassium phosphate buffer pH 7.0). After incubation for 10 min at the temperatures from 40–70 °C, the reaction mixture was boiled for 5 min to terminate the reaction. Then, 30 µL of mixtures were mixed with 170 µL chromogen solution, consisting of 1.47 mM DAOS, 2.2 mM 4–AA, and one unit/mL horseradish peroxidase in 20 mM potassium phosphate buffer (pH 7.0) and incubated for 5 min at 37 °C. Thermostability was investigated by incubating of the diluted enzyme at the temperatures from 40–70 °C for 10, 20, and 30 min.
The heat-treated enzyme solutions were immediately cooled in ice-bath, and the residual activity was assayed at 37°C. The relative activity of the enzyme was calculated in comparison with non- heated samples at room temperature. The experiments were performed in triplicate.
The effect of pH on the HOD activity was studied using Britton Robinson buffer (buffering range 5.0 to 9.0) with a concentration of 40 mM. For pH stability analysis, the enzyme solution was diluted with Britton–Robinson buffer (buffering range 4.0–11.0). After incubation at 37 °C for 30 min, the enzyme solution was assayed in the standard reaction mixture for residual HOD activity.
Study the effect of betahistine on the induction of HOD production
Histamine is relatively expensive, and for running the experiments and induction of HOD gene a cheaper but efficient source was highly desired. Therefore, we chose betahistine, a structural histamine analogue which is relatively cheaper and more affordable, to be assessed as the inducer. For this purpose, three experiments were designed and conducted as follow;
Experiment 1: Betahistine as sole carbon and nitrogen source
1% (v/v) of seed culture (OD600= 0.13) inoculated to 20 ml of BSM containing 0.1% Betahistine as sole carbon and nitrogen source. The media were incubated for 24 h, at 30 °C and 115 rpm. Grown cells were harvested from the culture by centrifugation at 4,000 g for 10 min at 4°C, washed twice with a large volume of 0.1 M phosphate buffer (pH 7.0), and resuspended in the same buffer for performing HOD assay.
Experiment 2: Betahistine as the sole nitrogen source
1% (v/v) of seed culture (OD600nm=0.13) inoculated to 20 mL of BSM containing 1% acetate as sole carbon and 0.1% betahistine as sole nitrogen source. The cells were grown and prepared for HOD assay analysis, as explained above.
Experiment 3: Betahistine as inducer agent
1% (v/v) of seed culture (OD600nm=0.13) inoculated to 20 mL of BSM containing 1% acetate as sole carbon and ammonium chloride as the nitrogen source in the presence of 0.1% Betahistine, and the cells were prepared for HOD assay as explained.
For negative control, the BSM medium containing 1% acetate as carbon and 0.1 % ammonium chloride as the nitrogen sources, was used. The BSM medium containing 1% acetate as carbon and 0.1 % histamine dihydrochloride as the nitrogen source was used for positive control.
{kind=link}