Plant materials and growth conditions. Wild-type aequorin-expressing transgenic Arabidopsis ColQ (Col-0 background) plants were kindly provided by Marc Knight36. The EMS-induced mutant population was described previously30. The 24 − 14 (subsequently referred to as mvk-1) mutant was backcrossed with the ColQ aequorin transgenic line three times (BC3F3). This backcrossed line was then used for phenotyping. Arabidopsis seeds were sown onto half strength Murashige and Skoog (MS) medium containing 1% (w/v) sucrose, 0.5% (w/v) phytagel, and 0.05% (w/v) MES pH 5.7. After 4 °C cold treatment for three days, the plates were placed vertically in a growth chamber (16 h light/8 h dark cycle, 22 °C, 100 µE cm− 2sec− 1 light intensity). For other experiments, 10-day-old seedlings or 3-week-old plants were grown in PRO-MIX soil (Premier Tech Horticulture) in a growth chamber (16 h light/8 h dark cycle, 22 °C, 70% humidity and 150 µE cm− 2sec− 1 light intensity).
Plasmid constructs and plant/protoplast transformation. Full-length MVK (At5g27450) and MKK3 (At5g40440) genes were amplified using gene-specific primers (Supplementary Table 2) and cDNA derived from wild-type plants. The PCR products were cloned into pDONR-Zeo (Invitrogen) or CloneJET (Thermo Fisher Scientific) vectors.
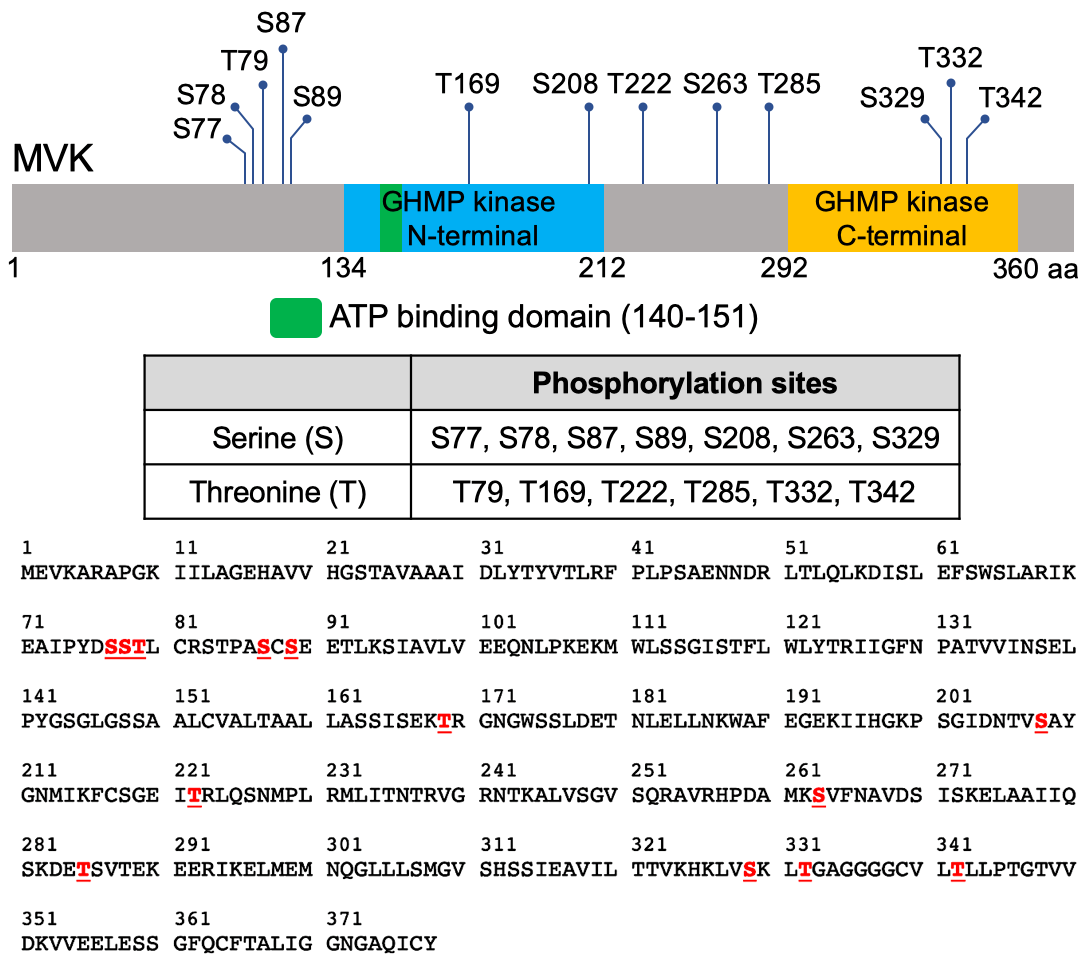
For constructs expressing recombinant proteins in E. coli, the cDNA of MVK and MKK3 were ligated into pET21a (Novagen) resulting in a His-tag fusion at the C-terminus of the protein. The P2K1 kinase domain and P2K1-1 kinase-dead in pGEX-5X-1 were previously described30. For cloning the various restriction enzymes (Supplementary Table 2) and T4 ligase (Promega) were used. MVK-S77A, S78A, T79A, S87A, S89A, T169A, S208A, T222A, S263A, T285A, S329A, T342A, and MKK3-kinase dead clones were generated by site-directed mutagenesis.
For Bimolecular Fluorescence Complementation (BiFC) assay, full-length coding sequences of MVK, P2K1, and MKK3 without stop codons were cloned into entry vector pDONR-Zeo and subcloned into pAM-PAT-35S::YFP, pAM-PAT-35S::YFPn, and pAM-PAT-35S::YFPc destination vectors through LR reaction66.
For co-immunoprecipitation assays in Nicotiana benthamiana, full-length CDS of MVK and MKK3 from the pDONR-Zeo vectors were cloned into pGWB17, and full-length CDS of P2K1 from the pDONR-Zeo vector was cloned into pGWB14 using LR reaction.
To generate constructs for the split-luciferase complementation assay in N. benthamiana, full-length CDS of MVK, P2K1, and MKK3 from the pDONR-Zeo vectors were cloned into pCAMBIA-GW-Nluc and pCAMBIA-GW-Cluc using LR reaction.
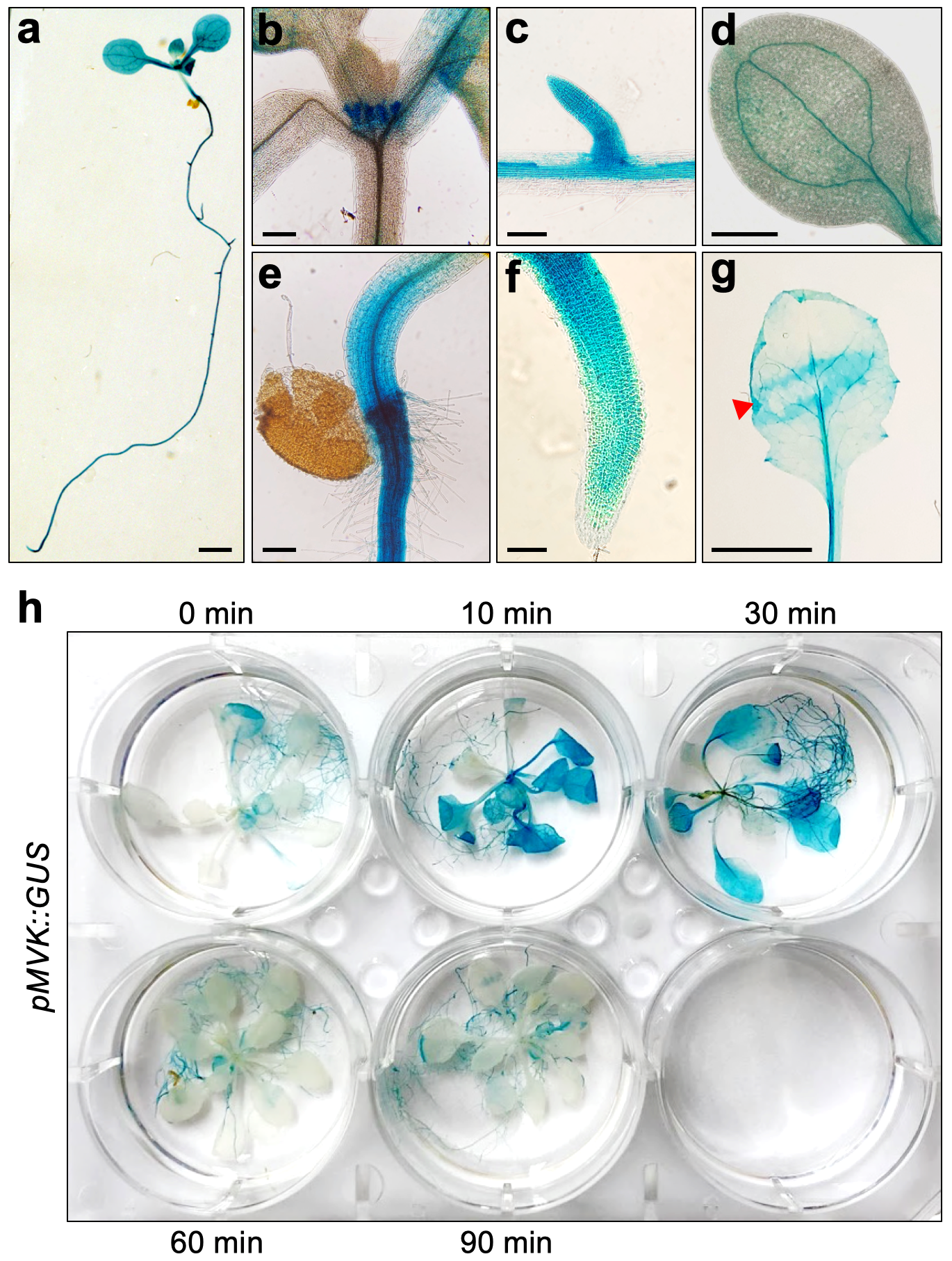
In order to generate stable transgenic Arabidopsis plants, the MVK promoter, a 1,350 bp region containing the 5’-UTR and encoding the first 17 amino acids, was cloned into pDONR-Zeo. To generate a fusion to the β-glucuronidase (GUS) reporter gene, the promoter region was subcloned into a plant binary vector pMDC162, generating the reporter construct pMVK::GUS.
The MVK promoter fragment (1,350 bp, see above) was fused with the MVK gene, followed by cloning into the binary vectors pGWB13 using LR reaction. MVK gene mutant clones containing different, mutated phosphorylation sites were generated by site-directed mutagenesis. Those fragments were cloned into the pGWB13 vector using LR reaction. These binary vectors were transformed into Agrobacterium tumefaciens GV3101 and used for transformation of Arabidopsis plants via the floral dip method67. The homozygous T3 lines were screened based on hygromycin resistance.
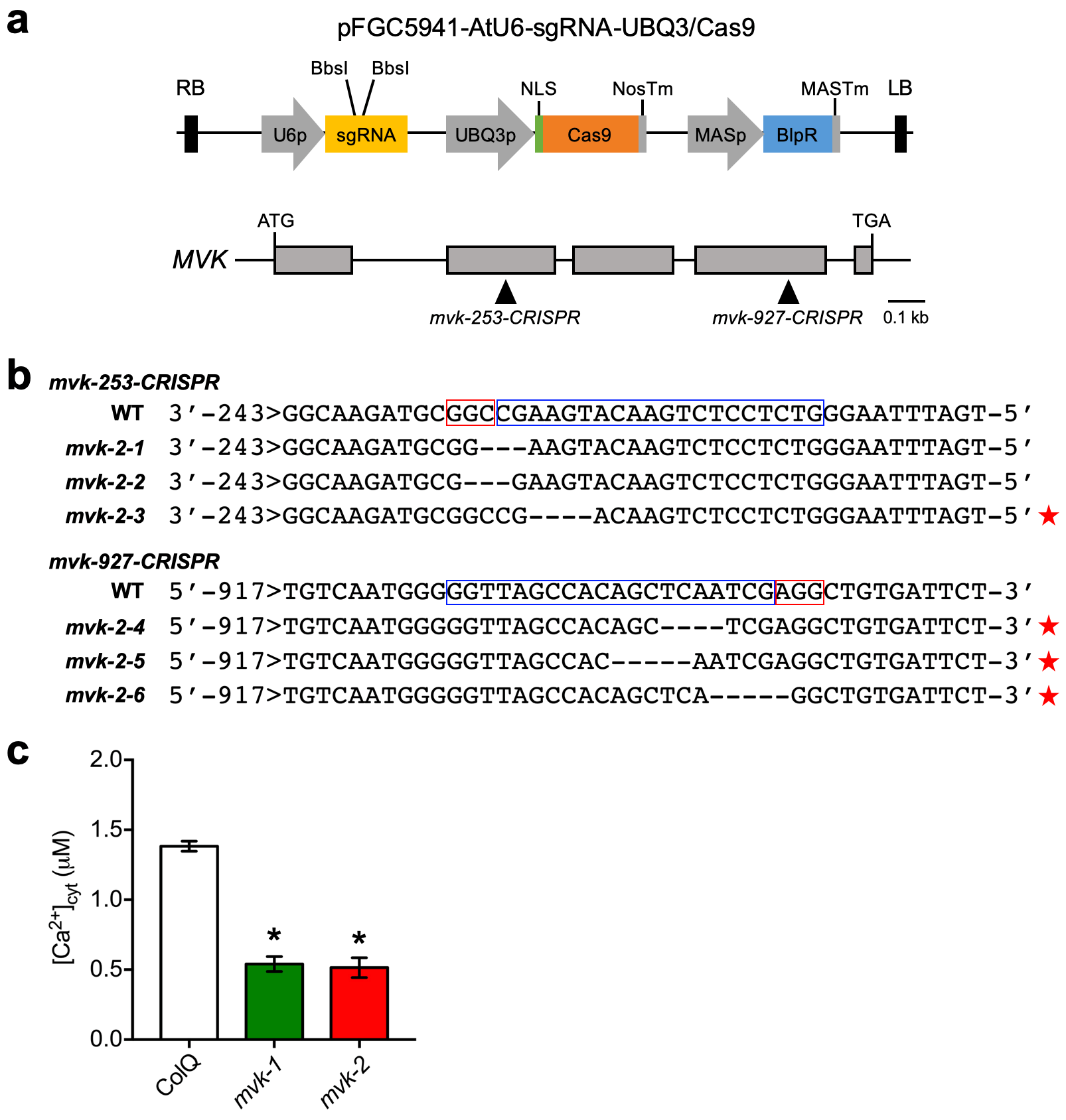
CRISPR/Cas9 editing was performed as previously described by Schiml et al68 (http://www.botanik.kit.edu/molbio/940.php). The Cas9 gene (UBQ3-Cas9-SK), driven by the UBQ3 promoter and the chimeric single guide RNA (AtU6–26-SK), driven by the AtU6–26 promoter was obtained from addgene (https://www.addgene.org/crispr/plant/). The bar gene, driven by mannopine synthase promoter of the binary vector pFGC5941, was used as a selection marker for Arabidopsis transformation. MVK specific single-guide RNA sequences were designed using the Zhang lab web-based tool: http://crispor.tefor.net/. Two gRNAs (MVK-253 and MVK-927) were used to create defined deletions within the exon of MVK gene. For each gRNA, a pair of DNA oligonucleotides (Supplementary Table 2) was synthesized and annealed to generate dimers. Subsequently, the annealed DNA was cloned using BbsI restriction sites into pAtU6–26-SK to create pSK-AtU6–26-gRNA, and sequence integrity was confirmed by Sanger sequencing (University of Missouri, DNA Core Facility). To obtain a functional Cas9 expression construct for targeted mutagenesis, pSK-AtU6–26-gRNAs were cut with EcoRI-SpeI, and UBQ3-Cas9-SK was digested with SbfI-SpeI. These 3 fragments were assembled into pFGC5941 by EcoRI-SbfI restriction digestion followed by ligation to obtain pFGC5941-AtU6- UBQ3/Cas9 construct. All primers used in this study are listed in Supplementary Table 2.
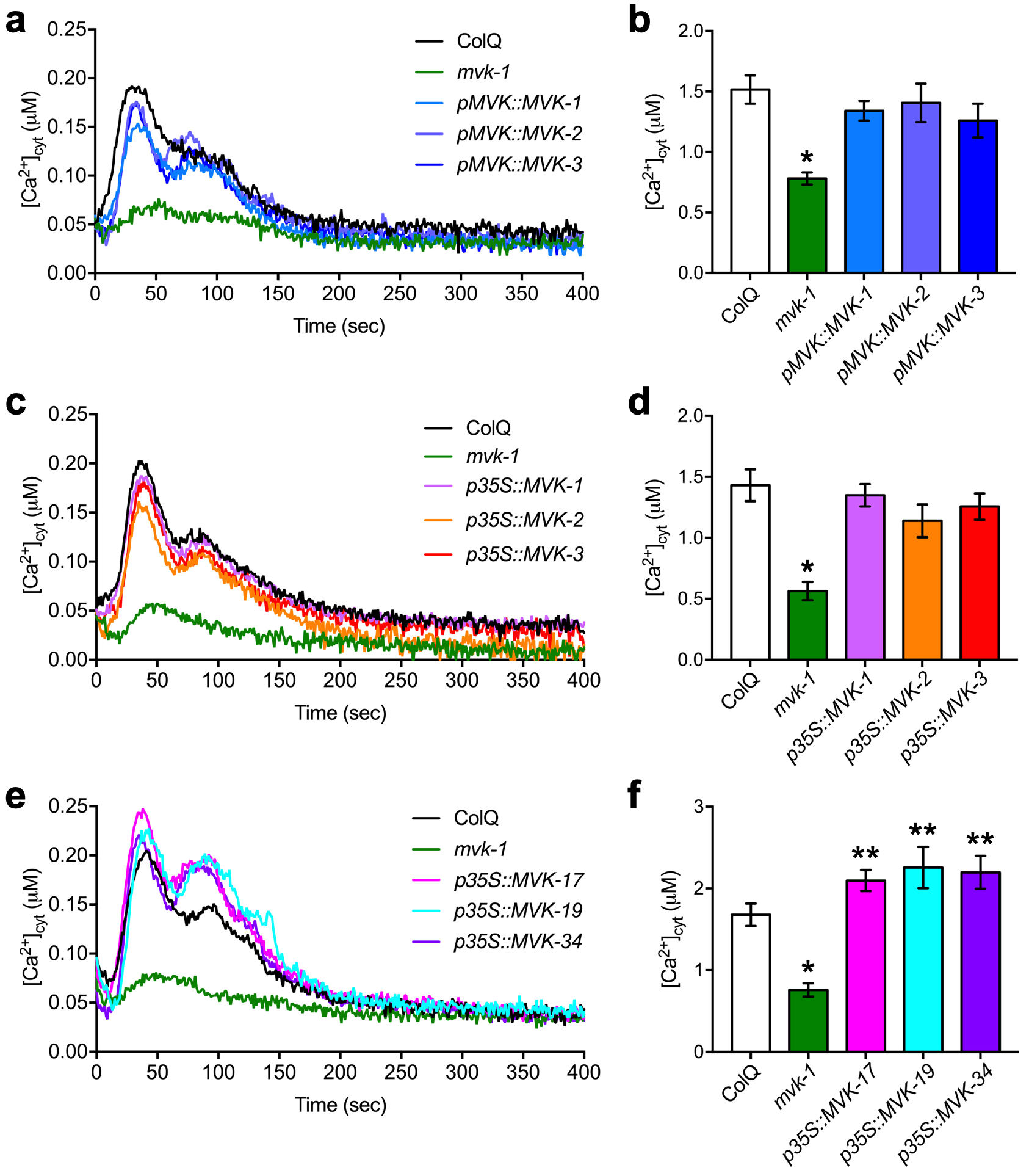
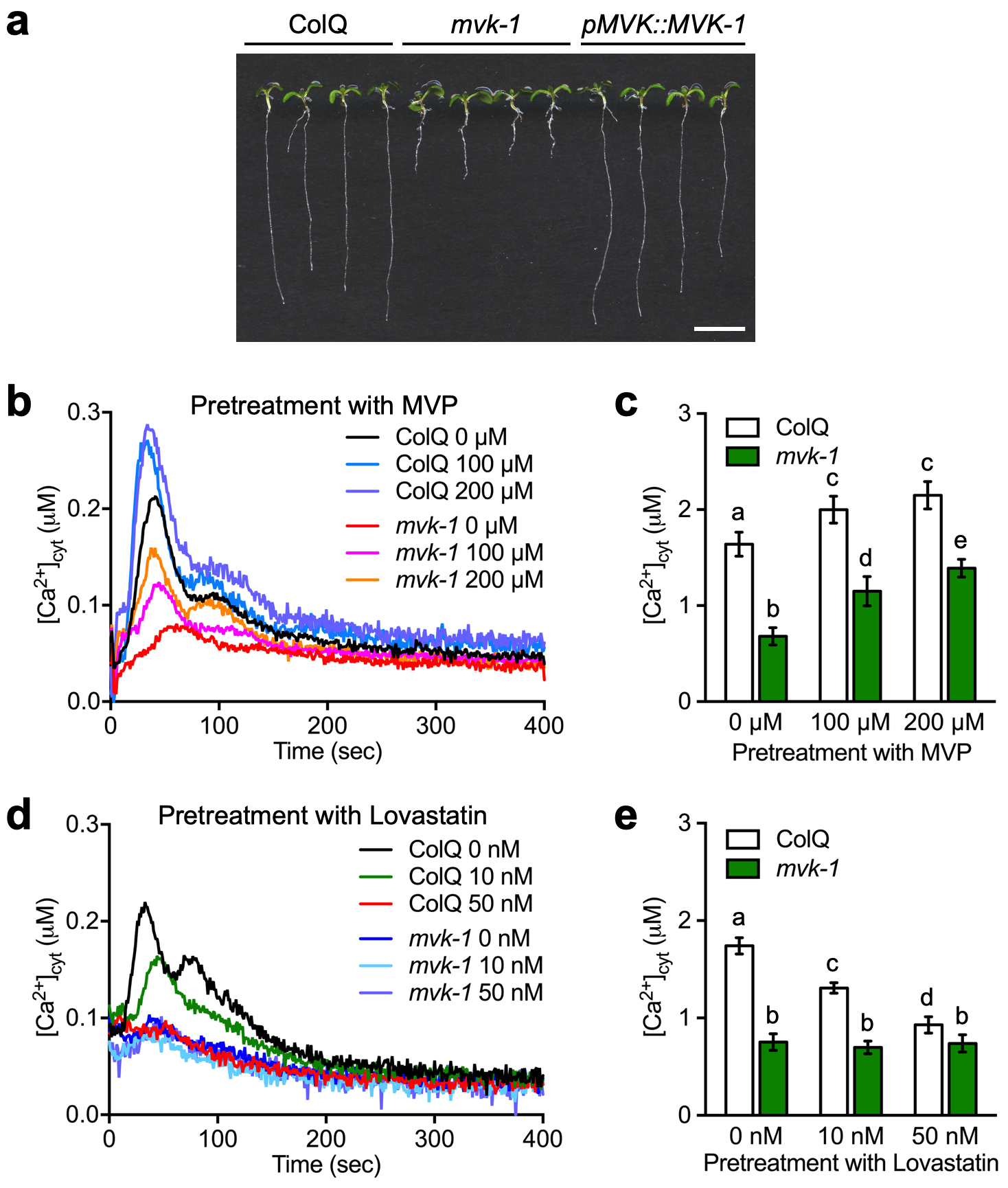
Cytoplasmic calcium assays. Assays were conducted as previously described30. Briefly, 5-day-old seedlings were individually transferred to a single well of a 96-well plate with 50 µl of reconstitution buffer containing 10 µM coelenterazine (NanoLight Technology), 10 mM CaCl2 and 2 mM MES pH 5.7, and incubated overnight at room temperature in the dark. Fifty microliters of nucleotides, abiotic and biotic elicitor treatment solution (double concentration) were applied in each well. The production of luminescence was monitored using an image-intensified CCD camera (Photek 216; Photek, Ltd.). One hundred microliters of discharging buffer containing 2 M CaCl2 and 20% (v/v) ethanol was used to estimate the remaining, unchelated aequorin. Photon counting data were converted into calcium concentration as previously described36.
EMS mutagenesis and mutant screening. In addition to the various p2k1 mutants identified previously by screening an EMS-mutagenized library derived from Arabidopsis thaliana expressing aequorin30, an additional 7 mutants were identified whose mutations did not map to the p2k1 gene. The mvk-1 mutant line was among these seven, which led to the identification of the mvk-1 mutant allele by map-based cloning and whole-genome sequencing. The response of the mvk-1 mutant plants to ATP was tested by applying an increasing concentration (10, 100, 500, and 1000 µM) of ATP. We also tested the mvk-1 plants for the specificity of their defect by applying a variety of known cytoplasmic calcium elicitors/treatments (e.g. 100 µM of ATP, ATPγS, ADP, ADPβS, AMP, Adenosine, GTP, ITP, CTP, TTP, and UTP; 100 nM of flg22, chitin, elf26, and pep1; ice-cold water; 5% D-glucose; 300 mM NaCl and mannitol).
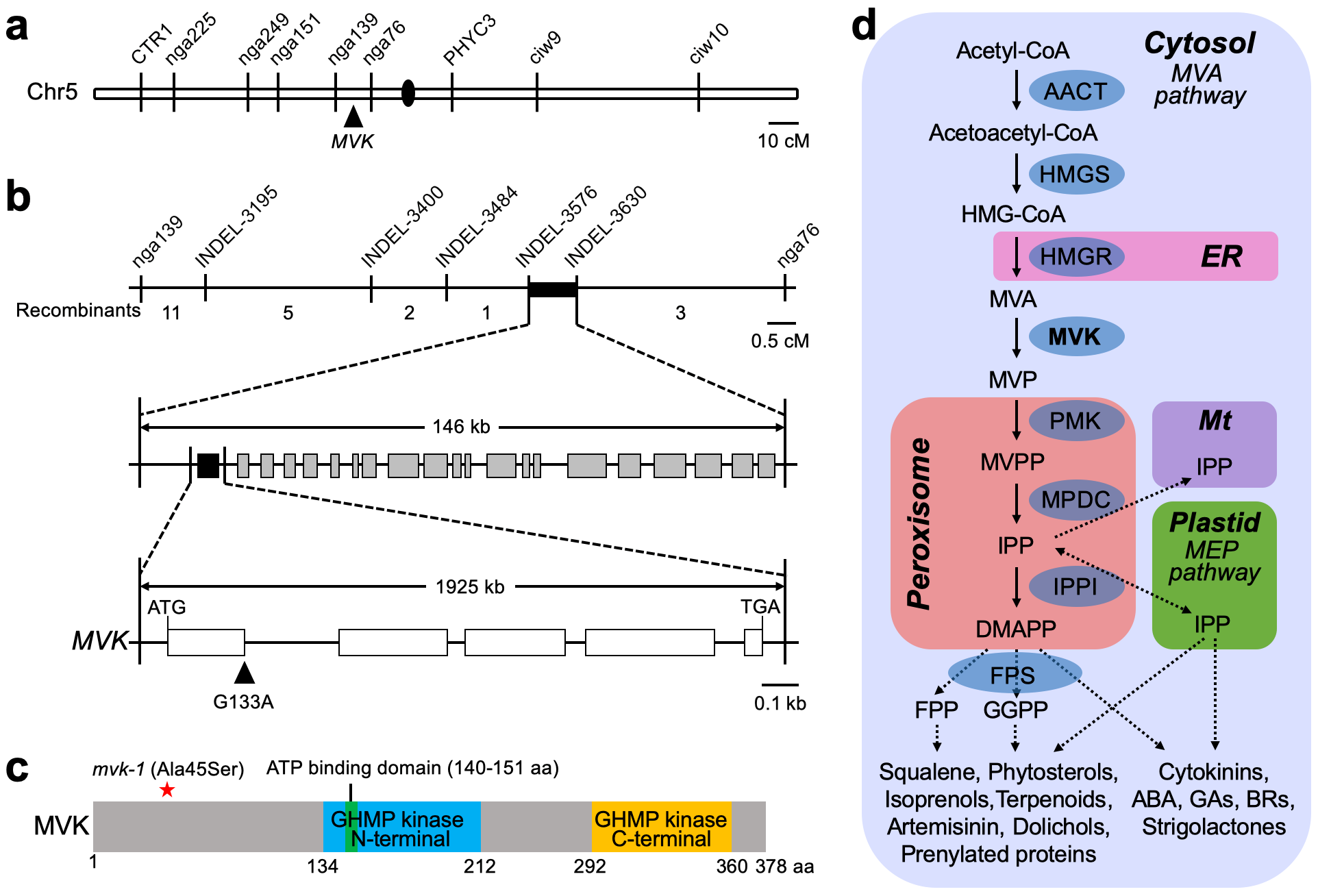
Map-based cloning. We generated a mapping population by crossing the mvk-1 mutant plants with A. thaliana Landsberg erecta ecotype plants, subsequently using the F2 generation for genotyping. The mutant phenotype was screened by monitoring the ATP-induced calcium response (via aequorin). Genetic mapping placed the mvk-1 mutation on the short arm of chromosome 5 as measured by co-segregation with the ATP response phenotype. SSR marker nga76 and nga139 on chromosome 5 were strongly linked to the mutant phenotype and additional INDEL markers were used for further mapping. We narrowed the mutant phenotype region in the map to an ~ 146 kb interval between INDEL3576 (two recombinants in 76 F2 populations) and INDEL3630 (one recombinant in 76 F2 populations). All the molecular markers are listed in Supplementary Table 2.
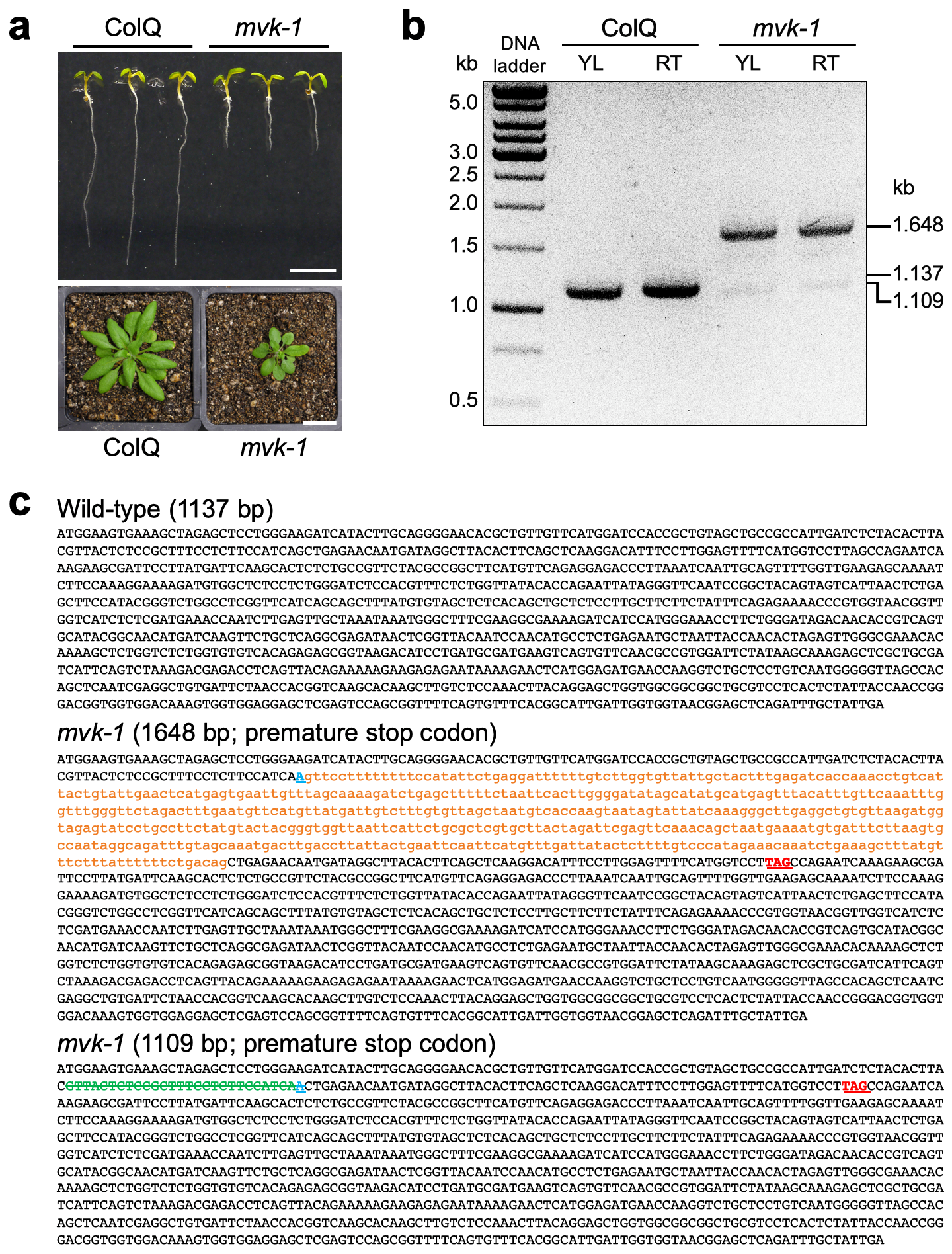
Whole genome sequencing. The mvk-1 mutant plants were backcrossed three times with wild-type ColQ and homozygous lines (BC3F3) were obtained for whole genome sequencing. As an internal reference, DNA from the ColQ originally used for EMS mutagenesis was also prepared for sequencing. Single leaves from 75 plants (3 weeks-old) were pooled and genomic DNA extraction was performed according to the manufacturer’s instructions (Qiagen, DNeasy Plant Mini Kit). The whole genome sequencing was carried out by the DNA Core Facility of the University of Missouri (https://dnacore.missouri.edu/ngs.html). Genomic DNA (3 µg) was sheared to 350 bp and used for DNA PCR-Free library preparation. Sequencing was performed on a Hiseq 2000 (Illumina) instrument with 100 bp single end reads (> 30 × coverage for all DNA samples using 1 × 100 run). Reads were quality trimmed using FASTX FASTQ Quality Trimmomatic version 0.3269. Reads were then aligned back to the TAIR10 version of the Arabidopsis genome using Bowtie version 270. Sam and output-pileup files were generated using Samtools version 0.1.771. The output-pileup files converted to the NGM emap file in Next generation mapping web tool (http://142.150.215.220/ngm/) and then analyzed single nucleotide polymorphisms as previously described72.
MAPK phosphorylation assay. Leaf discs from 3-week-old plants were incubated in 2 mM MES pH 5.7 at room temperature overnight. After treatment with 100 µM ATP for 0, 5, 10, 30, and 60 min, total protein was extracted with extraction buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.5% Triton-X 100, 1 mM DTT, 0.1 mM PMSF, and 1X protein inhibitor (Pierce) for 1 hour on ice. The extracted total proteins were mix with 5X Laemmli loading buffer containing 10% SDS, 50% glycerol, 0.01% bromophenol blue, 10% beta-mercaptoethanol, 0.3 M Tris-HCl pH 6.8, and heated in boiling water 5 minutes. The total extracted proteins were separated by 10% SDS-PAGE gel and detected by immunoblotting with rabbit anti-phospho-p44/p42 MAPK antibody (Cell signaling technology).
RNA isolation and quantitative real-time (qRT)-PCR. 10-day-old seedling plants were transferred into a 6-well plate and incubated in liquid MS medium in a growth chamber overnight. Samples were collected after treatment with 100 µM ATP, and total RNA was extracted using a RNeasy Plant Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA concentration was measured after Turbo DNA-free DNase (Ambion) treatment, and 1 µg RNA was used for first-strand cDNA synthesis using reverse transcriptase (Promega). The qPCR was performed using the PowerUp™ SYBR Green master mix (Applied Biosystems) following the manufacturer’s instructions. For data analysis, Rn data were extracted from ABI 7500 PCR machine and LinReg software (version 11.0) was used to determine Cq data and baseline. Transcript levels were normalized against the expression of the UBIQUITIN (UBQ) or SAND (At2g28390) gene. The gene specific primers used are listed in Supplementary Table 2.
MVK enzyme assay. Enzymatic activities were determined by following the procedure described by Reitzle et.al (2015). In brief, 2 µg of purified MVK-HIS protein was incubated with 2 µg GST-P2K1-KD kinase in reaction buffer (10 mM Tris-HCl pH 7.4, 5 mM MgCl2, 2 mM ATP, 100 µM Mevalonic acid). Reactions were kept at 30 °C for 10 minutes, and then terminated by adding an equal volume of acetonitrile. The samples were centrifuged for 20 min at 20,000 rcf and the supernatant was further processed for HPLC-MS/MS analysis. For quantification of mevalonic acid-5-phosphate, 50 µl internal standard working solution (containing 50 µM/L [13C, 2H3]-DL mevalonic acid-5-phosphate) were added to 50 µl enzyme assay sample, calibrator or quality control sample. After vortexing, the samples were evaporated to dryness under a gentle stream of nitrogen. Subsequently, 50 µl butanol–HCl (Sigma-Aldrich) was added to derivatize the carboxy acid group of mevalonic acid-5-phosphate (Sigma-Aldrich) and its internal standard to the corresponding butyl ester at 70 °C for 45 minutes. The samples were centrifuged for 2 minutes at 1500 rcf and evaporated to dryness. The residue was dissolved in 1000 µl water/acetonitrile (1:1) and further diluted 1:100 with water/acetonitrile (1:1). The diluted solution was loaded and analyzed by a Waters Alliance 2695 high Performance liquid chromatography (HPLC) system coupled with Waters Acquity TQ triple quadrupole mass spectrometer (MS/MS). The analytes were separated by a Kinetex C18 (100 mm x 4.6 mm; 2.6 µm particle size) reverse-phase column (Phenomenex). The mobile phase consisted of 10 mM ammonium acetate and 0.1% formic acid in water (A) and 100% acetonitrile (B). The gradient conditions were 0-0.5 min, 2% B; 0.5-7 min, 2–80% B; 7.0–9.0 min, 80–98% B; 9.0–10.0 min, 2% B; 10.0–15.0 min, 2% B at a flow rate of 0.5 ml/min. The ion source in the MS/MS system was electrospray ionization (EI) operated in the negative ion mode [M-H]− with capillary voltage of 1.5 kV. The ionization sources were programmed at 150 °C and the desolvation temperature was programmed at 450 °C. The MS/MS system was in the multi-reaction monitoring (MRM) mode with the optimized collision energy. The derivatized mevalonate-5-phosphate butyl ester was quantified with mass transition m/z 283◊96.
GUS staining and imaging. For detecting GUS activity, 10-day-old or 3-week-old plants were incubated in histochemical staining buffer (100 mM NaPO4 pH 7.0, 10 mM EDTA, 0.1% Triton X-100, 1 mM K3Fe(CN)6, 1 mM 5-Bromo-4-chloro-3-indoyl-beta-D-glucuronide) for 6 hours and then washed with 70% EtOH until tissue cleared. For the wounding treatment, rosette leaves were crushed using a hemostat forceps.
Co-immunoprecipitation assay. Agrobacterium tumefaciens GV3101 carrying the indicated constructs in infiltration buffer (10 mM MES pH 5.7, 10 mM MgCl2, 150 µM acetosyringone) was infiltrated into 3-week-old leaves of N. benthamiana. After 2 days, 200 µM of ATP and 2 mM MES (pH 5.7) were infiltrated into the same leaves. Total protein was extracted from pulverized (ground in liquid nitrogen) N. benthamiana leaf tissues using the following buffer: 50 mM Tris-HCl (pH 7.5), 250 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 1 mM DTT, 0.2 mM PMSF, 10% glycerol, 0.5% Triton-X 100, and 1X protease inhibitor (Thermo Fisher Scientific) by gentle rotation at 4 °C for 2 hours. The solution was centrifuged at 20,000 g for 10 minutes at 4 °C. The supernatant was transferred into a new tube and 1 µg anti-Myc (Sigma-Aldrich) was added, and incubated overnight with end-to-end shaking at 4 °C. Subsequently, 25 µl protein A resin (GenScript) was added for 4 hours, spun down and washed five times with washing buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, and 1X protease inhibitor. After washing, the resin was eluted with 50 µl 1X SDS-PAGE loading buffer and the eluent heated in boiling water for 10 minutes. The proteins were separated by 10% SDS-PAGE gel electrophoresis and detected by immunoblotting with anti-HA-HRP (Sigma-Aldrich; dilution, 1:1000).
Split-luciferase complementation imaging assay. Agrobacterium tumefaciens GV3101 containing the indicated constructs were incubated in infiltration buffer (10 mM MES pH 5.7, 10 mM MgCl2, 150 µM acetosyringone) for 2 hours in dark and subsequently used to infiltrate into 3-week-old leaves of N. benthamiana for 2 days before the LUC activity measurement. 1 mM D-luciferin (Goldbio) was sprayed 1 time onto the leaves, and then kept in the dark for 7 minutes to allow the chlorophyll luminescence to decay, the luminescence was monitored using a CCD camera (Photek 216; Photek, Ltd.).
Bimolecular fluorescence complementation assay. The subcloned YFP protein fusion constructs were co-transformed into Arabidopsis protoplasts by polyethylene glycol (Sigma-Aldrich) for transient expression73 and then incubated in a 23 °C chamber overnight in the dark. The fluorescence signals were monitored using a Leica DM 550B Compound microscope with Leica DFC290 color digital camera. FM 4–64 dye (Fisher scientific) was used to stain the plasma membrane for reference.
In vitro kinase assay. 2 µg of purified GST, GST-P2K1-KD or GST-P2K1-1-KD kinase was incubated with 1 µg MVK-HIS (WT, S77A, S78A, T79A, S87A, S89A, T169A, S208A, T222A, S263A, T285A, S329A, T332A, and T342A) as substrate in a 20 µl reaction buffer (20 mM Tris-HCl pH 7.5, 10 mM MgCl2, 5 mM EGTA, 100 mM NaCl, and 1 mM DTT, 2 mM ATP, and 10 µCi radioactive [γ-32P] ATP) for 1 hour at 30 °C. The reaction was stopped by boiling for 5 minutes with 5x SDS loading buffer. After electrophoresis in 12% SDS-PAGE, the gel was exposed for 12 hours for autoradiography. The proteins within the gel were visualized with coomassie brilliant blue. Myelin basic protein (MBP) (Sigma), GST, and MKK3-KD were used as controls. Experiments were repeated independently three times.
MVK phosphorylation site identification by LC-MS/MS. 20 µg of purified GST-P2K1-KD kinase was incubated with 10 µg MVK-HIS as substrate in a 200 µl reaction buffer (20 mM Tris-HCl pH 7.5, 10 mM MgCl2, 5 mM EGTA, 100 mM NaCl, and 1 mM DTT, 2 mM ATP) for 30 minutes at 30 °C. In-solution trypsin digestion of the 25 µl reaction was performed at 37 °C O/N and 1 µg protein was loaded to identify phosphorylation sites by Bruker timsTOF Pro. A 40 minutes chromatographic separation (LC) was performed using Thermo C8 Pepmap 100 trap column and Bruker C18 (75 µm x 250 mm) of 1.6 µm particle size (OD4-25075 C18A) served as the analytical column. MS/MS were acquired within 100–1700 m/z using PASEF (Parallel Accumulation-Serial Fragmentation)74 10 frames per 1.27 sec cycle (total 100–120 MS/MS). Automatic script (Bruker Data Analysis v5.1) was applied to convert RAW data to MGF file. Thermo Scientific Proteome Discoverer v 2.2 software running Sequest HT was used to search Arabidopsis proteome within NCBI database. Searches were conducted with tolerance of 50 ppm on the precursor and fragment ion mass tolerance of 0.1 dalton, and fixed modification of carbamidomethylation (C), variable modifications of oxidation (M) and phosphorylation (STY). Two biological replicate experiments were conducted.
Immunoblot assay. Total protein was extracted from 10-day-old Arabidopsis seedlings by homogenization in extraction buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 1 mM DTT, 0.2 mM PMSF, 10% glycerol, 0.5% Triton-X 100, and 1X protease inhibitor (Sigma-Aldrich) for 4 hours on ice. The samples were centrifuged at 20,000 rcf for 10 minutes at 4 °C, then the extracted total proteins were mix with 5X Laemmli loading buffer containing 10% SDS, 50% glycerol, 0.01% bromophenol blue, 10% beta-mercaptoethanol, 0.3 M Tris-HCl pH 6.8, and heated in boiling water 5 minutes. The total extracted proteins were separated by 12% SDS-PAGE gel and detected by immunoblotting with anti-HA-HRP (Roche, dilution 1:2000).
Metabolomic analysis. Polar and semi-polar metabolites from 10-day-old Arabidopsis wild-type and mvk-1 mutant roots were extracted following protocol75. Flash frozen pooled root samples were lyophilized and ground to a fine power with a tissue lyser. 800 µl of methanol/water (80:20) was added to 30 mg of pulverized root tissue, and samples were shaken at 1100 rpm for 1 hour at 21 °C in a Thermomixer. Samples were centrifuged for 5 minutes and the supernatant was transferred to a new vial. All extracts were kept at -80 °C until metabolomic analyses.
Root extracts were analyzed with a high-resolution mass spectrometer (HRMS) Orbitrap Velos (Thermo Fisher Scientific) coupled to a Thermo Vanquish HPLC (Thermo Fisher Scientific) equipped with a heated electrospray ionization (HESI) source (Thermo Fisher Scientific). C18 (Hypersil Gold 150 × 2.1 mm, 3 µm particle size) reversed-phase column (Thermo Fisher Scientific) was used for the liquid chromatography at flow rate of 300 µl/min, while column compartment was maintained at 30 °C. HRMS was performed using Fourier transform mass spectrometry (FTMS) and full-scan mode at high resolving power (60,000 full width at half maximum). A mass range of 50-1000 m/z was acquired for each ionization mode. Samples were analyzed in both negative and positive ionization modes. Experimental blanks consisting of methanol/water (80:20) were injected every ~ 15 samples and used to determine the chromatographic background. A mixture of standards was analyzed every 40 samples to test for mass accuracy of the instrument and for further RT and m/z calibration purposes. One injection of methanol/water (80:20) was analyzed right after the standard mixture to avoid any alleged carry over and it was not used for background determination.
Obtained LC-MS RAW files were processed using MZmine 2 version 2.3876 (Supplementary Table 3) and a final dataset was exported to a CSV file (Supplementary Table 4). Using MZmine 2, metabolic features were assigned to specific metabolite identities using an in-house LC-MS library which contains over 600 typical metabolites from the primary and secondary metabolism of plants. Metabolite annotations were based on exact mass and retention time (RT) of the detected features corresponding to a second level of identification as detailed by the metabolomics standards initiative77. Metabolite annotation information for LC-MS data is detailed in Supplementary Table 5.
To ensure that interesting candidate metabolites were not thrown out during statistical analysis, peak intensity tables containing both previously annotated and unannotated peaks were submitted for missing data imputation using Metlmp1.2 (https://metabolomics.cc.hawaii.edu/software/MetImp/)78. Following missing data imputation, statistical analysis of the CSV files was done using MetaboAnalyst 4.0 (https://www.metaboanalyst.ca/)79. Data was normalized by median, log transformed, and pareto scaling was applied. Following data preparation, both volcano plot analysis (significance P < 0.05, FC > 1.5) and fold change analysis were carried out. Box-and-whisker plots were generated via MetaboAnalyst for further analysis. Fold change values were generated for the 7000 + peaks in the normalized data set. Fold change values for the same peak in the various treatments were grouped and analyzed for interesting patterns and flagged for identification. It was then attempted to identify peaks of notable significance – from both volcano plot and fold change analysis – using the mMass software to correlate m/z values to known metabolites using in-house libraries generated from the PlantCyc database (https://plantcyc.org/)80. m/z values were matched to candidate metabolites within 0.02 da.
Bacterial inoculation assay. The bacterial inoculation assay was performed with minor modifications as previously described34. 2-week-old Arabidopsis seedlings were inoculated in 50 ml of Pseudomonas syringae pv. tomato DC3000 Lux (OD600 = 0.002) bacterial suspension (0.025% Silwet L-77 in sterile water) for 2 minutes at room temperature. After removing the bacterial solution, the plants were incubated in a growth chamber. The first day after inoculation, the aerial part of seedlings was collected and sterilized with 70% ethanol for 1 minute followed by a rinsing step with sterile water three times. Bacterial growth was visualized and measured under a CCD camera (Photek 216; Photek, Ltd.). The homogenized seedling tissue was diluted from 10− 2 to 10− 6 and dropped onto King’s B agar plates containing rifampicin and kanamycin for 2 days. The bacterial colony forming units (CFU) were counted and analyzed in GraphPad Prism 7.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}