Bacterial strains and Ribotype Confirmation
All C. difficile isolates used in this study are listed in Table 1. C. difficile UNT 101-1 to UNT-110-1 were kindly provided by Dr. Curtis Donskey (Cleveland VA); UNT 008-1, UNT 210-1, and UNT 196-1 were obtained from the American Type Culture Collection (ATCC). The source of relevant characteristics of each isolate can be found in Table 1. Ribotypes were confirmed by running polymerase chain reaction (PCR) ribotyping with primers found in Bidet et. al. [47]. PCR fragments were analyzed in a Hitachi 3500xL genetic analyzer with a 36 cm capillary loaded with a POP4 gel (Applied Biosystems). The size of each peak was determined using Peak Scanner software (Applied Biosystems). A database was generated from the results of the capillary gel electrophoresis-based PCR ribotyping result of each strain (http://webribo.ages.at). An error margin of ±4 bp was incorporated into the analysis algorithm of the database [48].
Media
Sporulation medium (SM) contained 90 g Trypticase Peptone, 5 g Proteose Peptone no. 3, 1 g Ammonium Sulfate, and 1.5 g of Tris in 1 liter of distilled water. The pH was adjusted to 7.4 at 37o with 1 M NaOH. SM is a broth medium made according to what has been previously described [49].
TSA with 5% blood agar was made with 1L of distilled water (DI), 30 grams of TSB, and 15 grams of granulated agar with constant mixing over low heat. Once the granulated agar was dissolved, the mixture was autoclaved (20 minutes, 121 °C, 15 psi). Once cooled to approximately 50 °C, 50 mL of the medium was removed, and 50 mL of sterile defibrinated sheep blood (Remel, Lenexa, KS) was added and mixed into the medium. Approximately 12 mL of medium was then poured into petri dishes and cooled overnight to solidify and stored in a 4 °C refrigerator until used.
TGY-vegetative medium contained 5 g Tryptone, 5 g Yeast extract, 1 g Glucose, 1 g Potassium Phosphate, 15 g agar, and 1 liter of distilled water. This liquid-based medium was made according to what has been previously published [50].
Columbia horse blood agar with 0.1% sodium taurocholate was made by adding 869 mL of distilled water, in combination with 35 g of Columbia broth (Remel), and 15 g of Difco Agar, granulated (BD). The mixture was autoclaved (20 minutes, 121 °C, 15 psi). Once cooled, 70 mL of horse blood and 50 mL of a 20 mg/mL stock of sodium taurocholate, 10 mL of a 50 mg/Ml stock of cycloserine and 1 mL of a 15.5 mg/mL stock of cefoxitin were also added.
Preparation of C. difficile spore stocks
Spore stocks of each C. difficile strain were generated for use in the cellular adherence assay and the experimental animal models of CDI. These stocks were generated by growing each strain on 5% TSAb plates incubated at 37oC in anaerobic conditions for 7 days. Plate growth was collected in a 1X PBS solution containing 1% (V/V) Tween-80 (ST-80), and suspensions were washed 3 times in equal volumes of ST-80. Suspensions were incubated for 1 hour at 65 ± 2°C, washed with ST-80, and re-suspended in 4 mL of sterile nanopore water. Suspensions were then stored overnight at 4°C in order to promote the maturation of endospores for each strain. Spores were separated from vegetative cells and residual debris by density gradient centrifugation (10 minutes at 4,500 x g) with a 25% (W/V) HistoDenz solution. Spore pellets were washed 3 times with ST-80 and suspended in sterile nanopore water to a final volume of 2 mL. Spore stocks for each strain were stored at -80°C until used in in vitro or in vivo studies, and the numbers of organisms given for infection or used in in vitro studies were confirmed for each study.
Mouse C. difficile associated disease model
Female C57 BL/6 mice that were 7 to 8 weeks old were obtained from Charles River Laboratory and housed in sterile caging for the in-life portion of each study. Animals were randomly organized into groups of 20 (n=20) and placed on drinking water supplemented with a cocktail of antibiotics immediately upon arrival. These antibiotics and their concentrations were: Kanamycin (0.4 mg/mL), Colistin (850 units/mL), Gentamicin (0.035 mg/mL), Metronidazole (.215 mg/mL), Vancomycin (0.045 mg/mL) [23]. Animals were left on the antibiotic supplemented water for 5 days, and then switched to normal water for 24 hours. Mice were orally inoculated with 1 x 106 C. difficile spores, and clindamycin was administered subcutaneously at 10 mg/kg of body weight. Starting the day of infection, and each day after, approximately 0.1 – 0.2 g of feces was collected from cages to determine C. difficile counts and associated amounts of toxin A and B. Bedding was changed daily to ensure fresh feces were collected for analysis, and census of survivors were recorded daily for 14 days after infection. Feces were weighed before sterile 1x PBS was added to the recovered feces, this solution was then homogenized, and 1 mL was separated for each total CFU recovery, spore recovery, and toxin A and B expression. Viable cell counts, spore counts, and toxin expression were quantified as described in the Material and Methods. The homogenized solution separated for spore quantification was heated to 65 ± 2oC for 1 hour to facilitate the isolation of only spores, while the fecal matter separated for toxin expression was diluted approximately 100x - 500x for quantification. This allowed it to fall within detection range of the ELISA used to determine toxin concentration.
Hamster LD-50/Survival C. difficile associated disease models
Male Golden Syrian hamsters that were 6 to 7 weeks old were purchased from Envigo RMS Inc., and individually housed in sterile cages. Up to 30 hamsters were used in each study with 5 animals in each group that were orally inoculated with a designated spore titer of each strain. The animals were inoculated with 0.5mL of C. difficile spores from a spore preparation culture though oral gavage. The inoculation dose for all strains ranged from 800 – 30,000 spores/mL, and the exact titers chosen for each strain were based on previously conducted studies and observation of higher titers with non-epidemic and epidemic strains. Clindamycin was administered subcutaneously to each animal at 10 mg/kg per body weight approximately 24 hours after infection. Starting the day of infection, and each day after, approximately 0.1 to 0.2 g of feces was collected individually from each cage to determine C. difficile counts and associated amounts of toxin A and B. Bedding was changed daily to ensure fresh feces were collected for analysis, and census of survivors were recorded daily for 7 days after infection. Cecal fluid was collected from deceased hamsters for C. difficile enumeration and toxin A and B quantification. Feces were weighed before sterile 1x PBS was added to the recovered feces, this solution was then homogenized, and 1 mL was separated for each total CFU recovery, spore recovery, and toxin A and B expression. Viable cell counts, spore counts, and toxin expression were quantified as described in the Material and Methods. The homogenized solution separated for spore quantification was heated to 65 ± 2oC for 1 hour to facilitate the isolation of only spores, and the fecal matter separated for toxin expression was diluted approximately 100x - 500x for quantification. Cecal fluid was processed identically to the fecal samples, with the exception that they were not homogenized. This allowed it to fall within detection range of the ELISA used to determine toxin concentration.
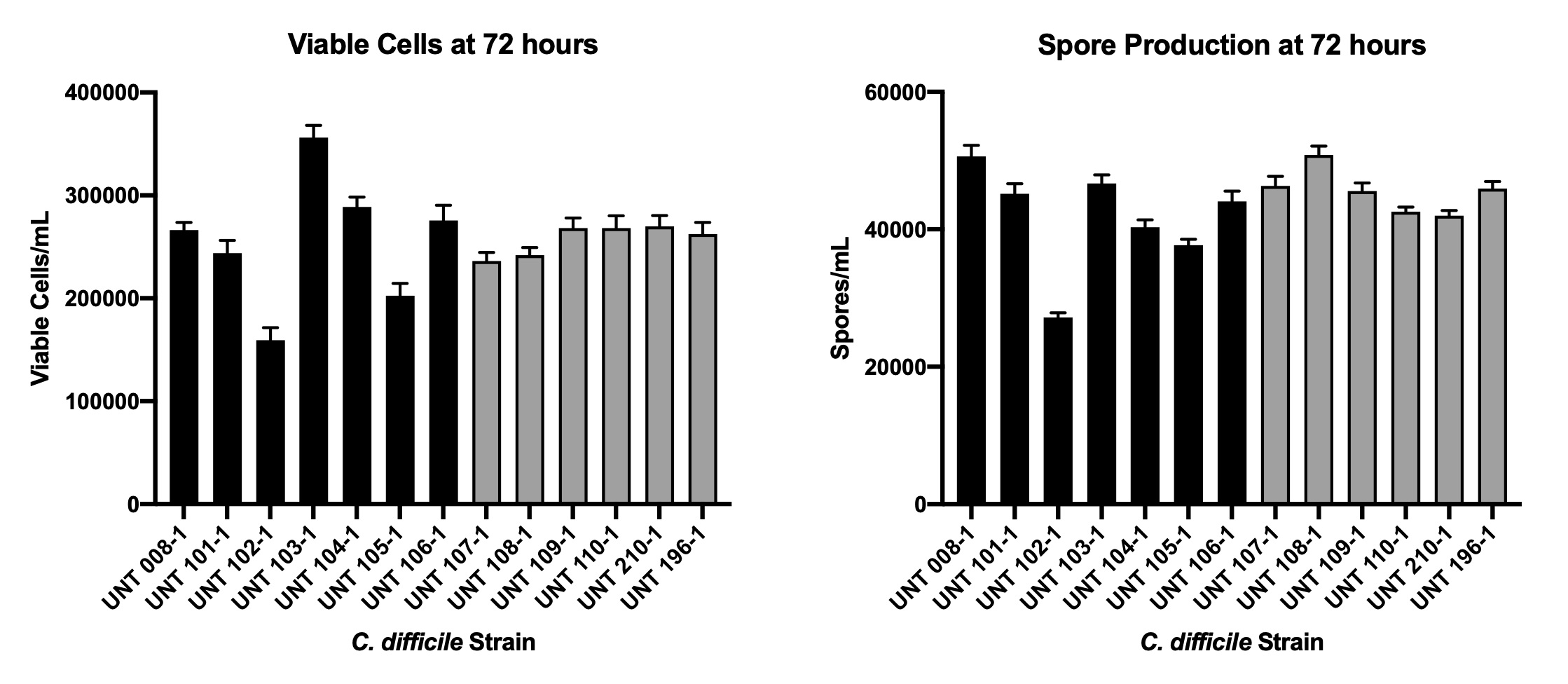
In vitro growth of C. difficile vegetative cells and spore formation
Plate growth of each C. difficile isolate was transferred into TGY-veg broth and anaerobically incubated at 37°C for 24 hours. TGY-veg associated growth for each strain was adjusted to an optical density of 0.1 (600nm) in either SM or TGY-veg broth, which were anaerobically incubated at 37°C. Samples from each broth culture were collected in triplicate every 24 hours through 72 hours of total incubation, and these samples were 10-fold serially diluted and plated onto Columbia horse blood agar. Additionally, a second sample from each culture were possessed for spore counts by incubating each sample in an equal volume of 200 proof ethanol for 30 minutes, and then incubating the samples at 65 ± 2oC for 1 hour. The ethanol and heat-treated samples were centrifuged, washed with PBS, and the spore-containing pellets were suspended in a volume of PBS equal to the original volume of the sample. Ethanol and heat-treatment at 65 ± 2oC were tested and sufficient to remove all viable vegetative cells during this stage. The spore suspension of each sample was 10-fold serially diluted and plated on Columbia horse blood agar supplemented with 0.1% sodium taurocholate. Both sets of plates were anaerobically incubated at 37°C for 48 hours and colony counts were used to calculate the vegetative CFU or spore counts per mL at each time point.
In addition to determining spore counts associated with each culture by counting the colonies recovered on agar media, the Schaeffer-Fulton endospore staining method was used to visually enumerate spores associated in 72-hour cultures of each C. difficile isolate. This was done by generating heat-fixed smears of samples taken from each culture every 24 hours on glass slides and staining with 0.5% (W/V) malachite green as each slide was being steamed for 5 minutes. Slides were counterstained with Gram’s safranin for 2 minutes in order to contrast vegetative cells from endospores and spores in each sample. The number of endospores and free spores were visually counted among 100 non-sporulating vegetative cells with a bright-field microscope at 1,000x total magnification, and the percentage of cells that had undergone sporulation was calculated for each C. difficile strain in triplicate at each 24-hour time point.
At the time of the viable cell quantification, 1.0 mL from the same sample vials were pipetted into 1.5 mL centrifuge tubes and centrifuged at 10,000 x g for 5 minutes. The supernatant was pipetted into a new 1.5 mL centrifuge tube and stored at -80°C until the quantification was performed.
Quantification of toxins
The levels of toxins A (TcdA) and B (TcdB) in fecal and culture samples were determined using an enzyme-linked immunosorbent assay kit purchased from tgcBIOMICS (Bingen, Germany). Samples were centrifuged at 10,000 x g for 5 minutes, and the recovered supernatants were diluted in kit supplied sample buffer. Toxin A and B concentration values for each sample were interpolated from standard curves generated for each toxin by non-linear regression analysis.
In vitro C. difficile adhesion assay
The Caco-2 cell line (ATCC HTB-37) and the C2BBe1 cell line were purchased from the ATCC. The Caco-2 cells were cultured in Eagles Minimal Essential Medium (EMEM) supplemented with 20% (V/V) fetal bovine serum (FBS), which was heat-inactivated, and 2 mM L-glutamine. The C2BBe1 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 0.01 mg/mL human transferrin and 10% (V/V) FBS. Other than the use of different growth media, the cell lines were grown and treated the same during the studies. The cells were grown at 37oC in an atmosphere of 5% CO2/95% O2, and spent media was replaced every other day until the cells reached 80-90% confluency. Caco-2 or C2BBe monolayers were removed from the growth flask with trypsin and transferred into 12-well tissue culture plates, which were placed into ncubator for 2 days, 37oC in 5% CO2/95% O2, to allow the cells to adhere to the wells.
To prepare for the assay, four aliquots of prepared C. difficile spore suspension of were washed twice by centrifugation and resuspended in PBS. For the adhesion assay, non-supplemented EMEM or DMEM replaced the medium currently in the wells containing the Caco-2 and C2BBe1 cells at least 1 hour prior to the assay, and C. difficile spores were seeded at a concentration of roughly 5 x 103 spores per well in triplicate. A negative control with PBS containing no bacteria was also added to additional wells in triplicate. Plates were incubated at 37oC in 5% CO2/95% O2 for 3 hours. Plates were removed from the incubator and the wells were washed twice with 1x PBS then the Caco-2 cell monolayer was detached from each well by adding a 1% (W/V) trypsin solution and anaerobically incubating the plates for 5 minutes at 37oC. The wells were, again, washed with 1x PBS, and the effluent was centrifuged at 8,000 x g for 5 minutes. Supernatants were discarded and each pellet suspended in 1mL of 1x PBS that was ten-fold serially diluted and plated onto Columbia horse blood agar. To enumerate spores the solution was plated on Columbia horse blood agar containing 0.1% sodium taurocholate.
Statistical analyses
Data were evaluated by One- or Two-way ANOVA with Tukey’s post-hoc test or unpaired Student’s t test. A p value ≤ 0.05 was considered statistically significant. Representation of survival rate against Log10 [daily dose]. LD50 values were calculated with the variable slope model (Y=100/ (1+10 ((LogEC50 – x) * HillSlope))) (Curve fitting, Prism 8, Graphpad Software, La Jolla, CA) and were compared for statistical significance using the extra sum-of-squares F test (p ≤ 0.05). Analyses were performed using Prism 8 software (Graphpad Software).
{kind=link}