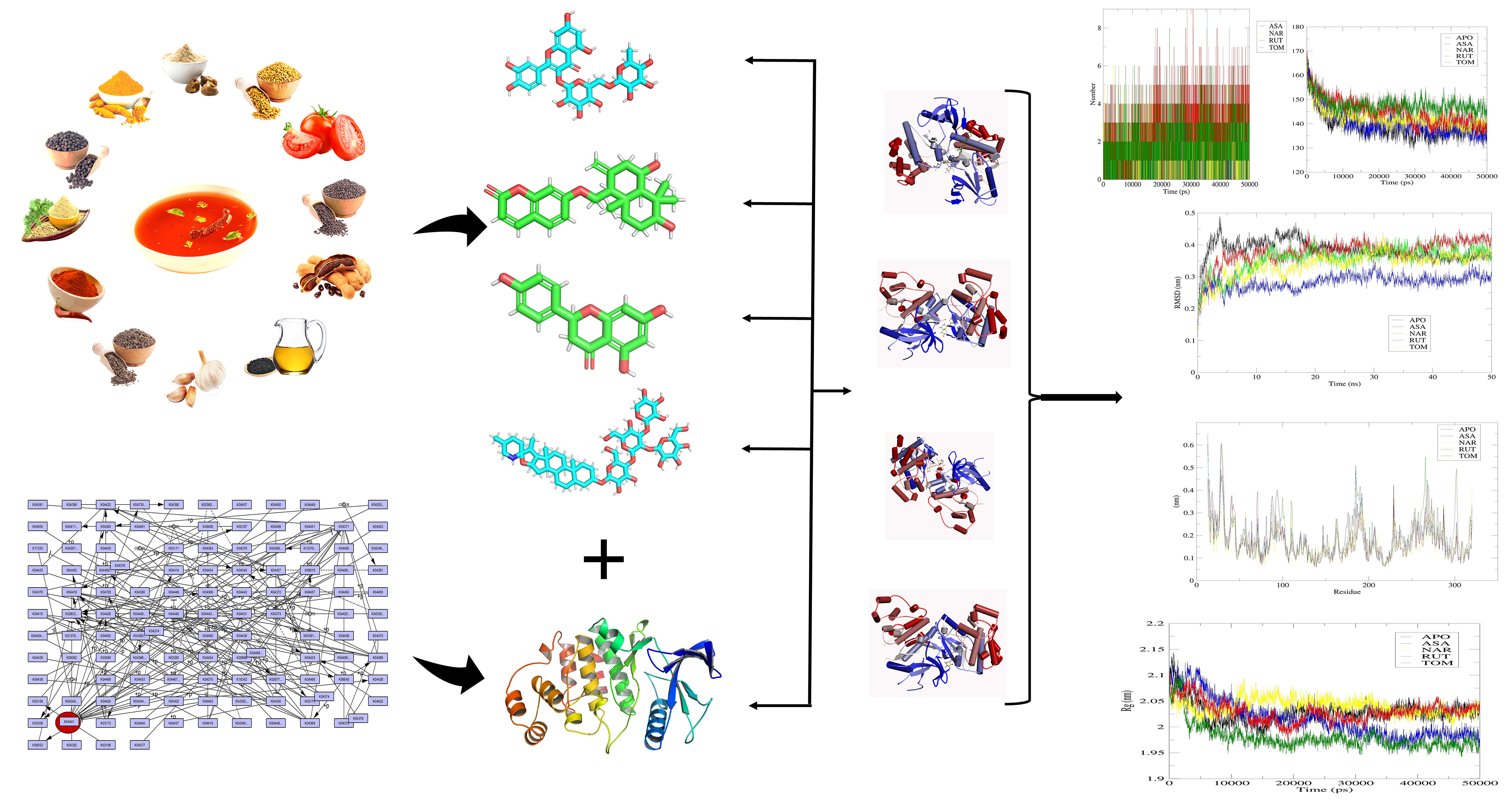
Bioactive compounds retrieval and preparation
The accessible bioactive components of the requested spices (tamarind, red pepper, black pepper, cumin seed, fenugreek, asafoetida, garlic, tomato, coriander, curry leaves, sesame oil, and mustard) were searched using IMPART database. From the database, a list of sixty-six important bioactive compounds were selected from the desired twelve spices depicted in Table 3.
Binding site identification
The crystal structure of MAPK (PDB: 7AQB) included 11 binding sites, according to binding site analyses. The protein's recovered binding site residue was shown in Figure 2. Molecular docking investigations were also conducted using the obtained complex structure of the binding sites. Grid generation in molecular docking research results in more reliable ligand posture scoring. As a result, we created a receptor grid for the selected MAPK protein based on the previously acquired binding site residues to achieve more precise scoring of our ligand poses. A receptor grid with a box diameter of X = 38.6666, Y = 62.5914, and Z = 31.9740 in angstrom (Å) was created and utilized further for molecular docking experiments.
Molecular docking
The optimum intermolecular interaction between the target protein and bioactive chemicals were investigated using molecular docking analysis. To analyze their binding capability, a specific number of bioactive chemicals (sixty-six) were docked against MAPK using PyRx tools AutoDock Vina. Twelve bioactive chemicals were shown to have a higher binding affinity (>-9 kcal/mol) with the target protein. The binding affinity of the bioactive compounds following molecular docking was found to be scattered, ranging from -3.50 to -10.60 kcal/mol, as illustrated in Fig. 3 and Table 3. The top four compounds (Assafoetidinol A (-9.80 kcal/mol), Naringin (-9.60 kcal/mol), Rutin (-9.80 kcal/mol), and Tomatine (-10.60 kcal/mol)) were chosen for future research based on their affinities with the active site aminoacid residues.
Interpretation of protein–ligand interactions
The interactions formed between the selected four ligands and MAPK has been visualized using BIOVIA Discovery studio visualizer tool. It was observed that compound CID: 2041593 (Assafoetidinol A) showed better interaction with binding affinity (-9.80 kcal/mol). Compound CID: 12041593 (Assafoetidinol A) formed four van der walls interactions with TYR (3.81 Å), GLU (3.57 Å), GLU (3.74 Å) and TRP (3.86 Å), two conventional and two carbon hydrogen bonds TYR (2.09 Å), ALA (2.10 Å) and GLU (3.58 Å), ASN (2.85 Å) respectively. Alkyl and Pi-Alkyl bond was also found at the position ARG (3.64 Å), ALA (3.75 Å), and PRO547 (3.67 Å), respectively showed in Figure 4 and Table 4. For the compound CID: 442428 (Naringin) it has been observed four hydrophobic and four hydrogen bonds with desired active site aminoacid residues of MAPK. Hydrophobic interactions with ALA (3.84 Å), LYS (3.76 Å), GLU (3.86 Å) and THR (3.75 Å), and four hydrogen bonds GLU (3.22 Å), LYS (3.11 Å), ASN (3.99 Å) and GLY (3.04 Å), respectively showed in Figure 5 and Table 4. In the case of CID: 5280805 Rutin exhibited four hydrophobic bonds with ARG (3.64 Å), GLU (3.59 Å), THR (3.71 Å) TRP (3.71 Å) and six hydrogen bonding with ARG (3.84 Å), ALA (4.03 Å), GLU (3.10 Å), ASN (3.20 Å), TYR (2.93 Å), GLU (3.44 Å) between active site aminoacid residues of desired protein MAPK, depicted in Figure 6 and Table 4. For, CID: 28523 (Tomatine), it has observed two hydrophobic bonding with TYR (3.97 Å), VAL (3.75 Å), and four hydrogen bonding with GLY (3.42 Å), ARG (3.25 Å), ASN (4.08 Å), THR (3.25 Å) between the target protein, and showed in Figure 7 and Table 4.
In silico Absorption, Distribution, Metabolism, and Excretion (ADME) prediction analysis
Absorption, Distribution, Metabolism, and Excretion (ADME) properties was assessed through SwissADME (www.swissadme.ch) webserver, which demonstrated that key bioactive compounds from rasam, i.e. Assafoetidinol A possessed better human intestinal absorption property. Naringin, Rutin and Tomatine have moderate absorption properties. In general, moderate intestinal absorption leads to the bioactive compounds of food (rasam) might be better consumed from the gastrointestinal tract upon oral administration. The higher number of H-bonds are possibly measured to be involved during protein ligand binding. From the result, the drug-likeness properties of four compounds showed better results (+0.55 Assafoetidinol A, and +0.17 for other three compounds) thereby relating with molecular properties, these four compounds were predicted to have better chances as a possible drug-relevant candidate with anticancer potential. The ADME properties like lipophilicity (dissolve in fats, oils and nonpolar solvents), water solubility and drug-likeness of the selected compounds have been investigated and presented in Table 5.
Analysis of toxicity
In silico toxicity prediction of the selected four compounds has been performed using ProTox-II web-based server. The server has identified drug-induced hERG toxicity, AMES toxicity, LD50, hepatotoxicity, skin sensitization, Tetrahymena pyriformis (TP) toxicity, and minnow toxicity which was listed in Table 6.
Molecular dynamics simulation
Although, protein–ligand docking was widespread and has successful use, it just gives the static view of the binding pose of ligand in active site of the receptor similar to photo image. Molecular dynamics (MD) must be employed to simulate the dynamics of atoms in the system as a function of time with integration of Newton’s equations of motions 38. MD simulations for 50 ns were carried out for the top four complexes obtained from the docking studies, that is 7AQB-ASA, 7AQB-NAR, 7AQB-RUT, 7AQB-TOM and unbounded apo form of the target MAPK protein (PDB ID: 7AQB) and their results were interpreted. To decipher the stability and fluctuations of these complexes, MD trajectories analysis was performed with the help of RMSD (Root Mean Square Deviation), RMSF (Root Mean Square Fluctuation), RG (Radius of gyration) and SASA (Solvent Accessible Surface Area) of receptor atoms.
RMSD is an important parameter to analyse the equilibration of MD trajectories and check the stability of complex systems during the simulation process. RMSD of the protein backbone atoms were plotted against time to assess its variations in structural confirmation. Initially, the 7AQB-ASA complex showed variations in backbone RMSD till 30 ns ranging from 0.15 to 0.44 nm. The stable conformation was attained in the time period between 21-50 ns with no considerable deviations in the values (Figure 8). 7AQB-NAR complex showed variations in backbone RMSD till 20 ns ranging from 0.17 to 0.43 nm. The stable conformation was attained in the time period between 21-50 ns with no considerable deviations in the values (Figure 8). The 7AQB-RUT complex showed variations in backbone RMSD till 30 ns ranging from 0.13 to 0.35 nm. The stable conformation was attained in the time period between 31-50 ns with no considerable deviations in the values (Figure 8). The 7AQB-TOM complex showed variations in backbone RMSD till 35 ns ranging from 0.14 to 0.43 nm. The first stable conformation was attained in the time period between 36-50 ns with no considerable deviations in the values (Figure 8). This clearly specifies that the protein underwent small structural changes in all the complexes during simulations.
RMSF is an another crucial parameter while examining the stability and flexibility of complex systems during simulation 39. RMSF was examined to analyse the change in behaviour of amino acid residues of target protein on binding to a ligand40,41. The RMSF values for Cα atoms of the protein were calculated and plotted with respect to the residues. In case of all complex, the amino acid residues showed minimal fluctuations throughout the simulation. The amino acids of MAPK which interacted with ASA during docking showed minimal fluctuation values during MD simulation viz. CYS28, GLY29, LYS185 and LYS229, with NAR it showed low fluctuation values during MD simulation viz GLY29 and LEU192, with RUT showed minimal fluctuation values during MD simulation viz. GLY29, ARG70, LYS229 and ASN269 and with TOM it showed moderate fluctuation values during MD simulation viz. GLY29, LYS185, SER189, TYR266 and PRO301 (Figure 9). These results reveal that binding of both the ligands actuated no major effects on the flexibility of the residues in the protein.
Further, Radius of gyration (Rg) of the complex systems were also analysed. Rg is the RMS distance of the atoms of the protein from the axis of rotation 41. It is one among the important parameter that represents the overall change in the protein structure compactness and its dimensions during the simulation42. Higher Rg values characterize the protein as less compact and flexible while low values depict the high compactness and rigidity39. Rg values of backbone atoms of protein were plotted against time to examine the changes in structural compactness. Binding of ASA decreased the backbone Rg values till 30 ns. In the time period between 31-50 ns there were no considerable fluctuations and almost constant value of ~1.98 nm was maintained. Till end, the Rg values were found to be in the range between 1.95-1.99 nm. Complete analysis revealed that, in the initial stage the trajectory had shown its peak value of ~2.12 nm. Later this high value was never displayed again which shows the stability of protein in the complex (Figure 10). Binding of NAR decreased the backbone Rg values till 15 ns. In the time period between 16-45 ns there were no considerable fluctuations and almost constant value of ~2.04 nm was maintained. Till end, the Rg values were found to be in the range of 2.02-2.05 nm. Complete analysis revealed that, in the initial stage, the trajectory showed its peak value of ~2.09 nm. Later, this high value was never displayed again which shows the stability of protein in the complex (Figure 10). Binding of RUT decreased the backbone Rg values till 31 ns. In the time period between 32-50 ns there were no considerable fluctuations and almost constant value of ~2.03 nm was maintained. Till end, the Rg values were found to be in the range of 2.00-2.05 nm. Complete analysis revealed that, in the initial stage, the trajectory exhibited its peak value of ~2.10 nm. Later, this high value was never displayed again which shows the stability of protein in the complex (Figure 10). Binding of TOM decreased the backbone Rg values till 10 ns. In the time period between 11-50 ns there were no considerable fluctuations and almost constant value of ~1.96 nm was maintained. Till end, the Rg values were found to be in the range of 1.94-2.00 nm. Complete analysis revealed that, in the initial stage, the trajectory had shown its peak value of ~2.10 nm. Later, this high value was never displayed again which shows the stability of protein in the complex (Figure 10). The complete interpretation revealed that both the molecules induced no major structural changes in the protein.
Moreover, analysis of Solvent Accessible Surface Area (SASA) for all the complexes was implemented. SASA is the substantial criterion to examine the extent of exposure of receptor to the surrounding solvent molecules during simulation39,43. In general, binding of ligand may induce the structural changes in the receptor and hence the area in contact with the solvent also may vary41. SASA values of protein was plotted against time to estimate the changes in surface area. For SASA complex, the trajectory showed decrease in the values till 15 ns. Except few time intervals, minute fluctuations were observed throughout the simulation period (Figure 11). The average SASA value was found to be ~138 nm2 and were in the range of 150-130 nm2. For NAR complex, the trajectory showed decrease in the values till 10 ns. Except few time intervals, minute fluctuations were observed throughout the simulation period (Figure 11). The average SASA value was found to be ~142 nm2 and were in the range of 149-134 nm2. For RUT complex, the trajectory showed decrease in the values till 10 ns. In the time interval of 11-28 ns, minute fluctuations were observed and from 29-34 ns a moderate fluctuation was observed (Figure 11). The average SASA value was found to be ~140 nm2 and were in the range of 154-133 nm2. For TOM complex, the trajectory showed decrease in the values till 10 ns. Except few time intervals, minute fluctuations were observed throughout the simulation period (Figure 11). The average SASA value was found to be ~148 nm2 and were in the range of 154-140 nm2. Overall, analysis revealed that the surface area of protein in both complexes were shrunken during the simulation.
To examine the binding affinity of the ligands with the target protein, the MD trajectories were analyzed to interpret the extent of hydrogen bond formation during the entire course of simulation and was depicted in Figure 12. SASA had formed good number of H-bonds with the receptor protein with a maximum of five bonds at several time frames indicating the stronger affinity towards the target. Consistency was maintained in forming almost two hydrogen bonds for the entire simulation time which signifies the stability of the complex. For the NAR complex, the consistency was maintained in forming three hydrogen bonds with maximum of six bonds at certain time periods. For rutin complex, the consistency was maintained in forming four hydrogen bonds with maximum of nine bonds at certain time periods. For the TOM complex, the consistency was maintained in forming two hydrogen bonds with maximum of nine bonds at certain time periods. This clearly signifies that the top phytochemicals have the stronger affinity with the target protein.
{kind=link}