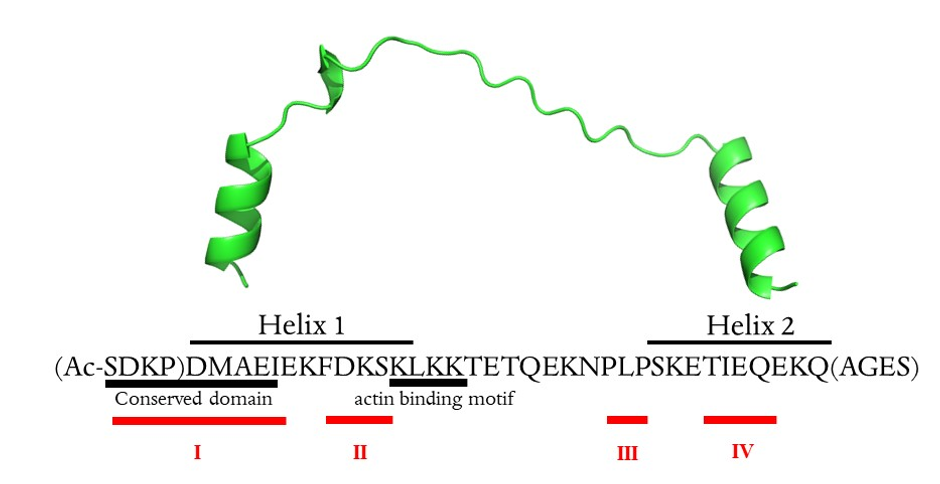
Thymosin β4 metal coordination.
Thymosin β4 is a 43 amino acid peptide with acetylated N-terminus. It is an acidic peptide (pI 5.1) due to the presence of 11 amino acids with carboxylic groups in the side chain, which are the potential metal binding sites 24. Moreover, nine lysine residues in the primary sequence have high affinity to transition metal ions 24. Thymosin β4 has no secondary structure in water, while in hydrophilic environment two alpha-helixes are formed in the 5–17 and 30–39 regions 25.
To our knowledge, no experimental or molecular dynamics study of the formation of essential metal complexes has been performed with Tβ4, despite there being numerous potential metal- binding sites. In order to investigate the metal binding sites in the Tβ4 peptide, we chose nuclear magnetic resonance (NMR) techniques, which deliver valuable structural data of metal complexes 26.
The interaction of iron ions with Tβ4 in water was investigated in a residue-specific manner by monitoring of 2D 13C-1H HSQC (heteronuclear single quantum coherence) NMR (Figure S1, S2, S3, S5, S6), focused on the aliphatic region. The overlay of the HSQC data (Figure S2, S3, S5 and S6) showed that metal/peptide interactions are nonspecific, and there is no structural change upon interaction with iron2+ nor iron3+ ions.
The presence of metal ions in the peptide solution leads to the lowering of the peptide signals. The comparative analysis of the peptide signal pattern with and without metal ion permits the isolation of amino acids affected by the interactions with the metal ions (Figure S4 and S7). In the thymosin solution containing iron2+ and iron3+ ions, eighteen and fifteen residues respectively (Table 1) showed a major drop in intensity. Since iron3+ is paramagnetic, we observed greater overall signal broadening of the residues, with the aforementioned residues showing major drops in intensity.
Circular dichroism (CD) structural analysis of free Tβ4 in water showed that the secondary structure is mainly unorganized, and only 20% could be associated to the helical form. The addition of iron ions to the peptide solution did not significantly change the secondary structure (Figure S9).
Table 1
Amino acid residues affected by metal/peptide interactions.
| Binding region | I | II | III | IV |
| Fe (II) | S1 | K3 | P4 | | M6 | | | K11 | F12 | D13 | S15 | L17 | T22 | P27 | L28 | P29 | S30 | T33 | I34 | | | A40 | S43 |
| Fe (III) | S1 | K3 | P4 | | M6 | I9 | | | F12 | D13 | S15 | | T22 | | L28 | | | T33 | I34 | Q35 | E36 | A40 | |
| Al (III) | | | P4 | D5 | M6 | I9 | E10 | | F12 | D13 | | | | | L28 | | | | I34 | Q35 | E36 | | |
In general, NMR data precision is limited by the presence of paramagnetic species and does not permit detailed coordination analysis with iron ions. For this purpose, Al3+ ions, which form similar stoichiometry and geometry complexes as Fe3+, were used to model Fe3+/ Tβ4 complexes.
The analysis of the overlay HSQC data (Fig. 1) showed that metal/peptide interactions are nonspecific, and there were no structural changes in thymosin upon interaction with Al3+ ions.
The chemical shift perturbation (CSP) was calculated considering the free peptide form and 1:5 (peptide:metal) as the maximum stoichiometry leading to the reduction of the peptide’s peak intensities and no further chemical shift changes. Only 48 isolated peaks were considered for calculating the CSP and intensity analysis. Analysis of the CSP plot (Fig. 2) showed that Al (III) ions interact with N-terminal residues of Tβ4. The residues that most likely interact with the metal ion are listed in Table 1.
Figure 2 shows that residues Ile9 and Gln35 presented noticeable changes upon interaction with Al3+, however the CSP value is too small to define the metal/peptide interaction as strong and with high affinity. The plot of signal intensities as a function of the Tβ4:metal ratio showed a consistent decrease for most of the residues in 1D (Fig S12) and in 2D (Figure S13). However, it is important to note that the intensity of 1D spectra is directly proportional only to the population, while the 2D spectra intensity is modulated by many different factors.
The diffusion NMR data at the different metallopeptide ratios showed the presence of only one species with a diffusion constant 1.25e10 m2/sec.
The NMR data analysis showed that metal ions interact with numerous amino acid residues, and four metal binding sites could be distinguished within the Tβ4 structure (namely I-IV, Table 1). The binding modes within four metal binding sites were modelled with the density functional theory (DFT) calculation.
Tβ4 contains 11 acidic residues: eight glutamate and three aspartate residues 1. According to Pearson’s hard and soft acids and bases (HSAB) principle, hard metal ions (Fe3+ and Al3+) show preference for binding hard bases, such as the negatively charged carboxylate side chains of glutamate and aspartate amino acids. Although there can be other donors to hard metal ions, such as backbone carbonyl and amide groups of proteins, by virtue of the partial covalent nature of the coordination bond, these donors are less competitive. Indeed, we recently demonstrated, from a thermodynamic perspective, that the preferred binding sites of Al3+ in proteins are the negatively charged side chains of amino acids rather than the backbone carbonyl and amide groups 28.
The chemical shift perturbation of the NMR data showed that the main changes in chemical shifts upon Al3+ addition arise in two different areas of the protein: within the N-terminal segment (metal binding sites I and II) and the C-terminal segment (metal binding sites III and IV).
The conformational analyses were prepared in order to build 3D models of the Al3+-Tβ4 complex and obtain some reliable initial configurations of this hard metal ion bound to Tβ4. According to the literature, Tβ4 in water solution exists mainly in a disordered state and it is unclear whether secondary structure elements such as extended helices in the N- and C-terminus of the protein arise upon interaction with other partners such as actin 23. Thus, in the starting structure, the Tβ4 sequence was consider “unfolded”, without main secondary structure elements (helices and sheets).
Three major binding modes for the Al3+-Tβ4 system were obtained (Table 2): first the metal interacting with both the N- and C-terminus of the protein (Tβ4N − C); second with the metal bound to the N-terminal and in the middle of the protein (Tβ4N − mid); third with the metal bound only in the N-terminus of the protein (whereas the rest of the sequence remains highly disordered; Tβ4N − N). Interestingly, all the three binding modes featured four negatively charged residues (side chains of either ASP or GLU) with a net charge of -4 surrounding the metal center, a situation that is coherent with the one proposed by Dudev and Lim 29, who stated that the optimal number of acetate-type donors in a metal binding site is equal to q + 1, where q is the charge of the metal, when first coordination shell residues are taken into account.
In Tβ4N − C binding mode, the metal is bound to GLU8 and GLU21 in a bidentate fashion, and to ASP5 and GLU35 by a single oxygen donor (Fig. 3A). The six oxygen donors coordinate Al3+ through an octahedral geometry, with O-Al-O angles ≥ 150 degrees. During the simulation, after 3µs the carboxylate side chain of GLU35 coordinates the metal with both oxygen donors whereas the side chain of GLU21 switches to a monodentate binding mode. The original state is then restored after 4 µs up to the end of the simulation. Overall, the TB4N − N protein-metal complex remains stable for the entire course of the simulation, and no other residues nor water molecules enter the first coordination shell of the metal.
The Tβ4N − mid binding mode is characterized by Al3+ ions coordinated to the side chains of ASP2 and ASP5 in a monodentate fashion, and to the side chains of GLU10 and GLU24 in a bidentate fashion (Fig. 3B). Also in this case there is no change in the coordination pattern of Tβ4N − mid during the entire simulation time of 5µs.
Finally, the Tβ4N − N binding mode displayed an octahedral geometry, in which the first coordination shell of the metal is filled by one oxygen donor from the side chains of ASP5 and GLU8 and two oxygen donors from the side chains of ASP2 and GLU10 (Fig. 3C). The metal center maintains this binding mode for the whole simulation.
Structural characterization revealed that Tβ4N − N displays the highest degree of disorder less compaction rate with the average root mean square deviation (RMSD) and radius of gyration (Rg) of 1.07 and 1.29, respectively, compared to Tβ4N − mid (0.89 and 1.11, respectively) and Tβ4N − C (0.92 and 1.15, respectively) as reported in Table S1. This is coherent with the fact that, in the case of Tβ4N − mid and Tβ4N − C, the N- and C- termini are closer due to their respective metal binding modes that involve residues either in the medial or distal region of the protein, as also highlighted by their shorter end to end distance compared to TB4N − C (Table S1). The average amount of intramolecular hydrogen bonds is similar for Tβ4N − C and Tβ4N − N and slightly higher for Tβ4N − mid (Table S1); most populated H-bonds and salt bridges for each binding mode are reported in Table 2.
As can be deduced from Table S2, all three metal-Tβ4 complexes exist mainly as random coil structures; the prevalent structural motifs are bends and turns, consistent with the disordered nature of Tβ4. Interestingly, Tβ4N − C and Tβ4N − mid display a similar β-sheet content (16.8 and 17.6 %, respectively), whereas Tβ4N − N shows a much lower content of this structural motif (2.5 %). On the other hand, the latter binding mode displays a higher helicity propensity compared with the other two, in particular it features 94.1 % of the 310-helix motif. Such a situation could be due to the fact that the metal binding activity of Tβ4N − N is confined to the N-terminal segment of the protein, while the remaining sequence is highly disordered and may increase the propensity to form helical patterns in its C-terminal regions.
Table 2
Percentage of hydrogen bond and salt bridge occupancies during the simulations. bb: backbone, sc: side chain.
| TB4N − C | TB4N − mid | TB4N − N |
| Donor | Acceptor | Occ. (%) | Donor | Acceptor | Occ. (%) | Donor | Acceptor | Occ. (%) |
| GLU35-Nbb | THR33-Osc | 27.6 | LYS38-Nbb | GLU35-Obb | 26.0 | THR22-Nbb | LYS19-Obb | 23.8 |
| LEU17-Nbb | LYS14-Obb | 25.8 | LEU28-Nbb | ASN26-Osc | 28.2 | GLN23-Nbb | LYS18-Obb | 22.2 |
| LEU28-Nbb | ASN26-Osc | 36.6 | ILE34-Nbb | LYS31-Obb | 26.7 | GLU21-Nbb | LEU17-Obb | 23.4 |
| THR33-Nbb | GLN36-Obb | 32.0 | LYS31-Nbb | LEU28-Obb | 21.1 | ILE9-Nbb | MET6-Obb | 53.0 |
| LYS31-Nbb | LEU28-Obb | 22.5 | | | | GLU8-Nbb | ASP5-Obb | 51.5 |
| | | | | | | LYS38-Nbb | GLU35-Obb | 20.0 |
All three binding modes are quite stable across the 5 µs time scale. We also observed that Tβ4 binding to aluminium could affect the overall secondary structure pattern of the thymosin. Since both glutamate and aspartate residues are the preferred acidic groups of hard metals, the three binding modes characterized herein could fit either Al (III) or Fe (III) metal ions.
Thymosin β4 and ferroptosis
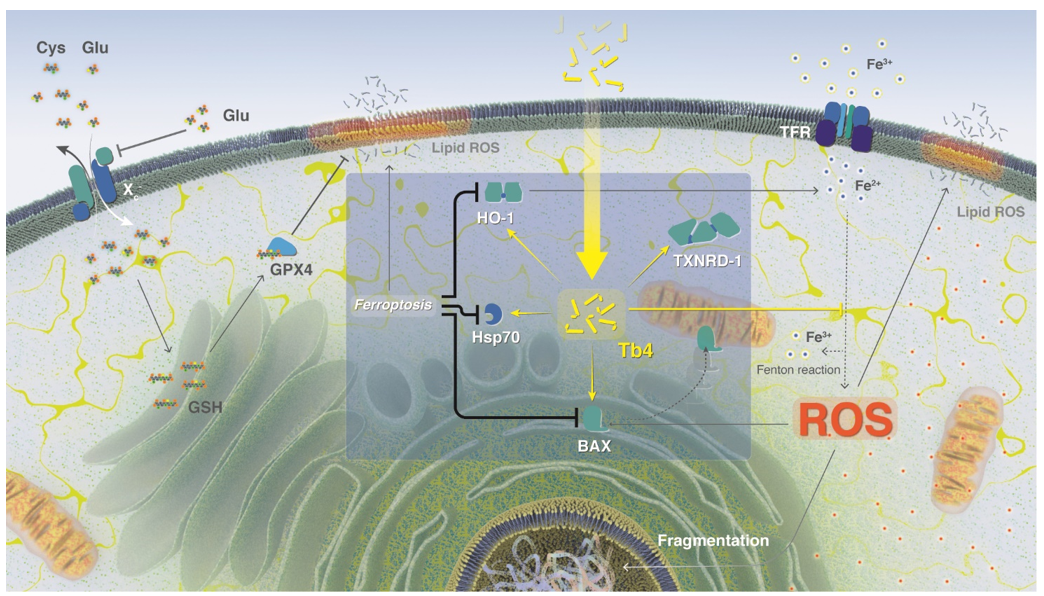
Iron is an essential metal ion with a fundamental role in diverse cellular processes, such as DNA synthesis, proliferation, cell cycle regulation, and the function of proteins containing iron–sulphur clusters 30. Iron–sulphur cluster-containing proteins include enzymes that contribute to maintaining genomic stability, as well as respiratory function 30. Indeed, rapidly reproducing cancer cells re-programme iron metabolism in ways that result in net iron influx 9,30. Iron is a highly reactive metal due to its redox potential, and when present in a free ion form, it contributes to the production of reactive oxygen species (ROS) in the Fenton reaction (Scheme 2). Oxidative and iron-dependent cell death with neurodegenerative consequences was previously described in epilepsy, stroke, and other trauma situations 31–33. Importantly, increased levels of iron were found in the central nervous system of patients with neurodegeneration 6. Surprisingly, elevated iron concentrations in cancer cells do not lead to cell disruption, but the exact mechanism of cell defense remains unknown.
Macrophages, together with hepatocytes, play key roles in iron metabolism by mediating iron storage and recycling 34. In hereditary haemochromatosis (HH), a representative disease that causes iron overload, hepatocytes are the first site of iron accumulation 35,36. Recent studies 37,38, showed that pathological iron overload in mouse macrophage cell line J774 induces ferroptosis and subsequent cell destruction. Therefore, we used J774 cells as a model to study the importance of macrophages in iron metabolism 39, and their consequent susceptibility to ferroptosis.
Ferroptosis can be induced in vivo in rat hippocampal slices and primary oligodendrocyte models as well as in ischemia-reperfusion injury models, by high concentrations of extracellular iron or glutamate, or by depletion of extracellular cysteine. In vitro, ferroptosis can be induced by physiological conditions (e.g., high extracellular iron or glutamate) or small molecules (e.g., Erastin), which blocks system Xc− - mediated Cys2 import 2 (heterodimer composed of solute carrier family 7, member 11 (SLC7A11) and SLC3A2; Scheme 2), while glutamate receptors are not involved in ferroptosis 40. Erastin and glutamate are termed type I ferroptosis inducers that inhibit cysteine uptake by the cysteine/glutamate antiporter (system xc−), enabling glutathione synthesis (Scheme 2), and thereby lowering antioxidant defenses 4.
Glutamate-induced oxidative stress leads to lipid peroxidation-mediated ferroptosis (Scheme 2) and is a major contributor to neurodegenerative pathologies 41. In recent studies, L-Glutamate (3h, 5mM) mimicked in vitro the consequences of stroke and neurodegenerative pathologies in cell cultures 42,43. The addition of iron enhances glutamate induced cell death 41, while inhibition of ferroptosis prevents glutamate-induced cell death in organotypic hippocampal slice cultures 4. Importantly, glutamate activity can be inhibited by iron chelators (e.g., Desferal) 4.
We induced ferroptosis in J774 cells in separate experiments by the addition of Erastin or glutamate and monitored the increase of free radicals with the BODIPY-C11 fluorescence marker (Fig. 4). 4,4-Difluoro-5-(4-phenyl-1,3-butadienyl)-4-bora-3a,4a-diaza-s-indacene-3-undecanoic acid (C11-BODIPY581/591) is a fluorescent probe (λex = 510 nm; λem = 595 nm) used for evaluating lipid peroxidation and antioxidant efficacy in model membranes, lipoproteins, biological fluids, and living cells 44, and has been used in ferroptosis studies 4.
After 24 h treatment with type I ferroptosis inducers (i.e., glutamate and Erastin), we observed a decrease in the green fluorescence light, while there was no change in fluorescence intensity in either the control nor the Tβ4 sample (Fig. 4). Notably, samples supplemented with glutamate (or Erastin), and successively with Tβ4 after 4h, showed the same fluorescence intensity after 24 h as the control sample.
Ferroptosis was primarily characterized by condensed mitochondrial membrane densities and smaller mitochondria, as well as diminished or absent mitochondria crista, and ruptured outer membrane 46. Recent studies divided mitochondrial morphological alterations into three categories according to the whether the mitochondria were fragmented or not, and whether they accumulated around the nucleus. Cells with a network of elongated mitochondria were assigned to category І, uniform-distributed fragmented mitochondria in cells were classified as category Ⅱ, and cells with fragmented mitochondria mainly accumulating around the nucleus were classified as category Ⅲ 46–49. Mitochondrial morphological changes and metabolic regulation, especially the energetic, iron and aliphatic acid metabolism involved in ferroptosis, were extensively described in a recent review 50.
For a more detailed characterization of glutamate-induced ferroptosis in J774 cells, including Tβ4 inhibition, we evaluated the morphological changes by TEM microscopy. The results obtained from this ultrastructural analysis are summarized in Fig. 5 (and Figure S19), 6 and 7, showing cells from different experimental groups. In the control group (Figs. 5A and 5B), the J774 cells appeared normal at the ultrastructural level. They were mainly characterized by a well-developed Golgi apparatus, rough endoplasmic reticulum, and the presence of numerous mitochondria. The nuclear compartment was separated from the cytoplasm by the double membrane of the nuclear envelope, and the cells appeared to be in interphase due to the presence of both heterochromatin and euchromatin. The presence of condensely packed chromatin and the absence of the nuclear envelope represented the main features of mitotic cells, which were often observed in control samples (Figure S19).
Administration of Tβ4 (Fig. 5C, D) did not appear to alter cell morphology: samples displayed structural integrity of all their cellular compartments and organelles including mitochondria.
Following glutamate (Fig. 6A and 6B) and Erastin (Fig. 7A and 7B) treatments, the cellular morphology of the J774 cells was dramatically altered. Specific ultrastructural alterations were mainly observed in the cytoplasmic compartment, where numerous mitochondria were found accumulating around the nucleus. These cellular organelles appeared drastically altered, showing mitochondrial membrane rupture and evident cristae disorganization. Moreover, the presence of lysosomal activity and disruption of the Golgi apparatus and rough endoplasmic reticulum indicated a poor cellular condition. However, none of these morphological alterations were related to an apoptotic form of cell death. Indeed, during our TEM investigation, no nuclear alterations were detected, and the nucleus appeared surrounded by a continuous nuclear envelope, displayed an evident nucleolus and well-organized chromatin, and no chromatin condensation or margination were detected in the J774 cells after either glutamate or Erastin treatments. Moreover, both Erastin and glutamate treatment seemed to induce cell detaching and a rounded cellular shape (Fig. 6A). Their cell surface appeared continuous without any signs of plasma membrane rupture or disorganization, although we did observe thin protrusions. Most of these appeared to be continuous tunnelling nanotubes connecting neighboring cells (Fig. 6A, asterisk) previously described as structures involved in intercellular communication 51,52.
Administration of Tβ4 in glutamate (Fig. 6C and 6D) and Erastin (Fig. 7C and 7D) treated samples seemed to gradually reverse the alterations described above. The cytoplasmic compartment showed reorganization of its cellular structures associated with an evident diminishing of lysosomal activity. Mitochondria showed partially reconstituted cristae and a continuous mitochondrial membrane. The rounded cell shape transitioned to a more flattened type.
Interestingly, in the glutamate and Erastin-treated cells we frequently detected the presence of tunnelling nanotubes (TNT) connecting neighboring cells (Fig. 7A, asterisk). TNT are considered dynamic structures whose formation and organization can be affected by different pathological conditions. It has been reported that oxidative stress may induce TNT formation in astrocytes and hippocampal neurons 53. Inflammation and even viral infections have been observed to spread via tunnelling nanotubes. And they may also be involved in the cell-to-cell transfer of signal transduction molecules. Fas ligand (FasL) involved in programmed cell death has been demonstrated to stimulate TNT formation, promoting propagation of cell death signals to neighboring cells 54. Our results suggest a possible involvement of the ferroptotic process in the cell surface reorganization and TNT formation, which may facilitate diffusion of the ferroptosis process between connected cells. Considering that tunnelling nanotubes are F-actin containing protrusions of the cell surface, Tβ4 could modulate ferroptosis not only by its metal binding sites, as described above, but also through its G-actin binding properties. Tβ4, as a major G-actin sequestering protein, could prevent tunnelling nanotube formation in Erastin and glutamate treated J774 cells inhibiting propagation of ferroptotic cell death signals to the connected cells.
These morphological results clearly show that glutamate and Erastin induce ferroptosis in J774 cells, while Tβ4 administration after 4 h can inhibit the process and reverse the critical cellular condition. The biochemical pathways involved in these processes were investigated by gene expression profiling.
According to Dixon2, ferroptosis could be defined as a sabotage mechanism, where the cell inhibits own-normal processes. However, ferroptosis is related to oxidative stress, and consequently the involvement of oxidative stress proteins is expected. Indeed, among thirty different genes studied in relation to ferroptosis55, many are involved in oxidative stress. In our study, we chose four genes, namely BAX (Bcl-2 associated X-protein), HSP-70 (Heat shock protein 07), which are involved in the oxidation process, but are poorly described in connection with ferroptosis; and HO-1 (hem oxygenase) and TXNRD-1 (Thioredoxin reductase 1) which were recently linked to ferroptosis.
Bax is a cytosol protein, which translocates into mitochondria when apoptosis occurs56 (Scheme 2). Jungas et al. 57 studied glutamate toxicity and regulation in neuronal development. Their studies showed BAX-activation mechanism dependent on glutathione, with downstream effect of glutathione depletion leading to the direct activation of BAX. Moreover, Bax and its transcript increase significantly in cerebral ischemia-injured neurons58,59 suggesting that glutamate could be involved the mechanism of BAX expression control.
Heat shock protein 70 (Hsp70) is a stress-responsive protein increasing under stress conditions, namely heat, hypoxia, and glucose deprivation. Increased Hsp70 concentration leads to higher stress tolerance and cytoprotection 60. Hsp70–mediated cytoprotective effects were studied in vitro in numerous cell/tissue types under changing environmental conditions of stress. Noteworthily, Hsp70 have significant cytoprotective effects under ischemic conditions. For instance, induction of Hsp70 (i.g. by enforced overexpression with viral vectors or drugs) protects brain cells against ischemic injuries 61–64. Oxidative stress and antioxidants can influence Hsp70 expression 65,66; while reduction in Hsp70 expression leads to higher ROS generation and mitochondrial protein oxidation67. Guo et al. 60 showed that Hsp70 regulates cellular redox status by changing the activities of the GSH-related enzymes, glutathione peroxidase and glutathione reductase, in response to hypoxic and ischemic stress. The switching of antioxidant enzyme activities could be critical mechanism mediating the enhanced cytoprotection afforded by enhanced expression of Hsp70. Despite numerous studies and different theories, the exact molecular mechanisms of Hsp70 action in ischemic conditions remains undefined
Heme oxygenase (HO)-1 metabolizes heme into biliverdin/bilirubin, carbon monoxide, and ferrous iron, and has been suggested to demonstrate cytoprotective effects under various stress-related conditions. HO-1 is commonly regarded as a survival molecule, exerting an important role in cancer progression, and its inhibition is considered beneficial in several cancers 68. HO-1 is often upregulated in tumor tissues, and its expression is further increased in response to therapies 69. The role of HO-1 in ferroptosis is extensively described in the recent review 68. Depletion of cellular glutathione has been shown to increase HO-1 gene transcription in the mouse motor neuron-like hybrid cells, NSC34 cells 70. The correlation between HO-1 expression and ferroptosis is unclear. In HT-1080 fibrosarcoma cells, Erastin induces a time- and dose-dependent increase of HO-1 expression 71. However, HO-1 also functions as a negative regulator in Erastin- and Sorafenib-induced hepatocellular carcinoma, since knockdown of HO-1 expression enhanced cell growth inhibition by Erastin and sorafenib 72.
The selenoprotein thioredoxin reductase 1 (TXNRD1 gene) is a cytoplasmic protein that decreases thioredoxin levels and protects against oxidative stress 73. Recently, it was shown that TXNRD1 is a strong negative modulator of ferroptosis susceptibility in pancreatic cancer cells 74. Moreover, a high-dose (25 mg/kg) of the anti-rheumatoid arthritis drug auranofin (AUR) upregulates hepcidin expression and induces ferroptosis and causes lipid peroxidation through inhibition of thioredoxin reductase (TXNRD) activity, in C57BL/6J mice and a mouse model of hemochromatosis (Hfe−/− mice) 75.
Here, we evaluated the levels of BAX, HO-1, Hsp70 and TRNRD-1 mRNAs (Fig. 8) under normal and ferroptosis conditions, which was induced by the extracellular overload of glutamate. In addition, we evaluated mRNA expression levels of these proteins in the presence of high extracellular concentrations of thymosin. Finally, we examined their mRNA levels in the cells treated with thymosin after 4 h of ferroptosis induction. We observed that glutamate overload led to the downregulation of all studied genes; conversely the presence of Tβ4 upregulated mRNA expression, particularly of HO-1. Notably, the downregulation of glutamate could be inverted by the successive treatment with Tβ4.
{kind=link}
{kind=link}
{kind=link}