Methods
Cell culture
The human trophoblast cell line HTR8/SVneo has been widely applied as an early invasion and migration model of extravillous cytotrophoblasts [6]. HTR8/SVneo, which was provided by American Type Culture Collection (USA), was cultured with Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS; Gibco, USA) at 37°C and 5% CO2.
Construction of stable cell lines
Plasmids containing AQP3 knockdown, AQP3 overexpression, or their respective negative control plasmids were purchased from Weijiang Biotechnology (China). Lentiviral packaging was performed in 293T cells according to the manufacturer’s instructions (Suzhou GenePharma, China). Lentivirus in the supernatant was collected to transfect cells. Strict phenotype selection was performed on stably infected HTR8/SVneo cells with 0–10 μg/mL puromycin (MPbio, USA) to use resistance as a screening index. Furthermore, cells were stably cloned in 0.5 μg/mL puromycin.
Total RNA extraction and RT-PCR
Total RNA was extracted according to the instructions of a TRIzol kit (Taraka Biotechnology, China), and measured with a spectrophotometer (Nanodrop 2000; Thermo Scientific, USA). cDNA was synthesized from total RNA (1 μg) using a fluorescent quantitative reverse transcription kit (Takara, Japan) and RT-PCR was performed by fluorescent quantitative PCR kit (Takara). PCR primer sequences were as follows: GAPDH forward 5ʹ-GAAGCTCATTTCCTGGTATGACA-3ʹ and reverse 5ʹ-GGGAGATTCAGTGTGGTGGG-3ʹ; AQP3 forward 5ʹ-ACCATCAACCTGGCCTTTGG-3ʹ and reverse 5ʹ-GGGGACGGGGTTGTTGTAG-3ʹ. The 2-ΔΔCT method was used to quantify relative expression of AQP3 mRNA.
Western blotting
Cells were collected and lysed with RIPA lysis buffer and phenylmethylsulfonyl fluoride (Beyotime Biotechnology, China) on ice for 30 min, quantified by bicinchoninic acid assay, and loaded for SDS-PAGE electrophoresis. After transfer to a polyvinylidene fluoride membrane, the membrane was blocked in 5% skim milk, sealed for 1 h at room temperature with shaking, and incubated with an AQP3 primary antibody (Abcam, UK; 1:1000) at 4°C. Finally, the membrane was incubated with horseradish peroxidase-labeled secondary antibody (1:20000) at room temperature for 1 h, and developed by enhanced chemiluminescence. Image Pro-Plus 6.0 software (Media Cybernetics, USA) was used to analyze gray values.
Cell proliferation/CCK-8 assay
HTR8/Svneo cells (100 μL; 1 × 105 cells/mL) in vector control, knockdown, and overexpression groups were added into 96-well plates in triplicate, and cultured at 37°C overnight. CCK-8 kit reagent (10 μL; Dojindo, Japan) was added into each well and incubated for 2 h, 3 h, and 4 h. Optical density at 450 nm of each well was detected each time point with a multifunctional microplate reader.
Flow cytometry assay
Annexin V-APC/7-AAD double staining was performed on HTR8/Svneo cells according to the instructions of an Annexin V-APC/7AAD Apoptosis Detection Kit [Multisciences (Lianke) Biotech, China]. Live, early apoptotic, and late apoptotic or necrotic cells were classified using flow cytometry (AccuriC6, Becton Dickinson, USA) and Flow Jo 7.6.1 software (FlowJo, USA).
Wound healing assay
HTR8/Svneo cells (1 × 106 cells/mL) were seeded in a six-well plate and routinely cultured in an incubator. When cells grew into a monolayer, they were treated with mitomycin for 1 h to inhibit cell division. Next, a sterile 10 μL-pipette tip was used to scrape cell culture plates. Scraped cells were washed twice with phosphate-buffered saline, cultured in serum-free medium, incubated in an incubator, and photographed at 0, 6, 24 and 48 h after scratching. Image Pro-Plus 6.0 was used to measure scratch depth at any five sites at the same time point to calculate migration rates, thus reflecting cell mobility and migratory capabilities.
Transwell invasion assay
A Transwell invasion system (8-μm, 24-well; Corning, UK) coated with Matrigel (40 μL; Becton Dickinson) was used. Briefly, 1 × 105 cells were suspended in DMEM without serum, and seeded in the upper chamber. DMEM containing 10% FBS was then added to the lower chamber and the plate was incubated at 37°C and 5% CO2. After 24 h, cells were fixed with methanol and stained with 0.1% crystal violet. The quantity of colored cells in five random visual fields was counted using an inverted microscope (Nikon, Japan).
Whole genome expression profile
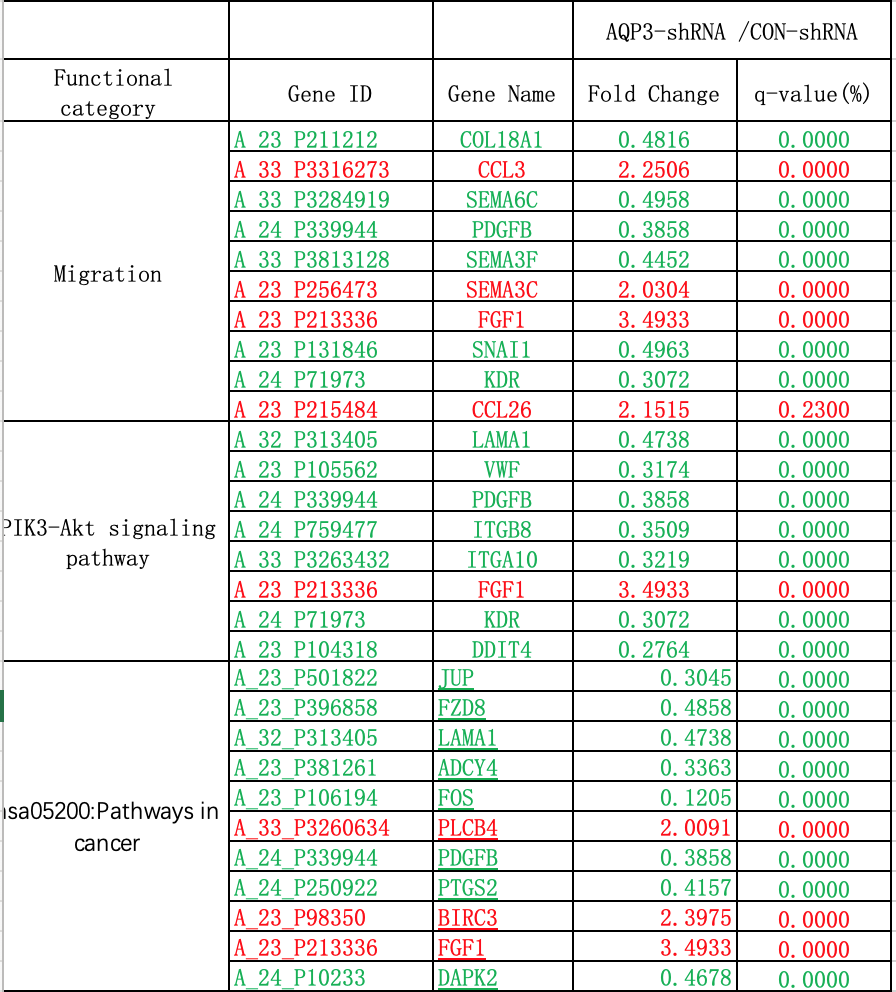
Gene expression profiles of AQP3-shRNA and CON-shRNA were analyzed by two-color gene expression microarray (Agilent Technologies, USA) according to the instructions of a Low Input Quick Amp Labeling Kit Two-Color (Agilent). Total RNA obtained in the extraction phase was used as a template, and the first strand of cDNA was reverse transcribed using T7 RNA polymerase. The second strand of cDNA was used as the synthesis template to perform in vitro transcription and promote generation of cRNA. An Agilent cRNA labeling kit was used to incorporate cRNA with Cy-3, which allowed purification and qualification of cRNA (Nanodrop 2000). After hybridization, washing, and chip scanning, data were extracted to perform bioinformatic analysis using Agilent Feature Extraction Software. Doing q-PCR verfication for FDGF-B,FOS and Snail1, which showed significantly decrease in the results of the gene expression profile experiment.
Statistical analysis
Data were analyzed by SPSS23.0 software (IBM, USA). Each experiment was performed in triplicate, and data were expressed as mean ± standard deviation (SD). Migration rate and invasion index were analyzed using a two independent-samples t-test. Proliferation and apoptosis rates were analyzed by analysis of variance. P < 0.05 was considered statistically significant.
{kind=link}