Study site, adult mosquitoes, larvae collection, and identification
We selected the study site in Manila City, the capital city of the Philippines. It is a highly populated and urbanized area that connects two major cities, Manila City and Quezon City. It consists of residential, commercial, and industrial infrastructure. We collected adults and larvae between June and September 2017, the rainy season and the peak time for dengue cases.
Adults were collected using commercially available mosquito light traps (Jocanima Corporation, Manila, Philippines). We set the mosquito trap for 48 hours either inside or outside the selected household premises (n = 428). The mosquito trap attracts the mosquito to enter a capture net by heat and CO2 transmitted by a strong current from the ventilator [20;21]. We surveyed water containers in each household (n = 428) and found 17 containers with Aedes larvae. The adults (n = 371) and larvae (n = 509) collected were morphologically identified as Aedes sp. using the keys published by Rueda et al. [22]. The adult and larval samples were preserved in RNAlater (Ambion, Invitrogen, CA) solution to keep their RNA and DNA intact and stored at −20℃ before nucleic acid extraction.
Using microsatellite data from a recent study [21], we were also able to confirm the species of 359 adult mosquitoes as Ae. aegypti. We had already used the adult DNA of 359 Ae. aegypti for population genetics study [21] and reused the data in this study. The microsatellite primers were species-specific for Ae. aegypti [23;24]. There was PCR amplification observed using the primers on the 12 Ae. albopictus samples.
DNA extraction, molecular identification, PCR amplification, and sequencing
We extracted DNA using Qiagen AllPrep DNA/RNA micro kits and Qiagen DNA Blood and Tissue DNEasy Kits© (Qiagen, Hilden, Germany) in adult (n = 371) and larval (n = 509) samples, respectively. DNA concentration and quality were measured using a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Wolbachia was detected using two molecular markers: wsp (610 base pairs) using the primer pairs wsp 81F (5′-TGGTCCAATAAGTGATGAAGAAAC-3′) and wsp 691R (5′-AAAAATTAAACGCTACTCCA-3′) [15], and 16S specific for Wolbachia (850 base pairs) with the primer pairs WolbF (5′-GAAGATAATGACGGTACTCAC-3′) and Wspecr (5′-AGCTTC GAGTGAAACCAATTC-3′) [25].
For both wsp and 16S gene amplification, we followed the protocol published in [7], in a final volume of 10 µl with 1 µl of the genomic DNA. We used the following components for the PCR reaction for both markers: 10x Ex Taq buffer, 25 mM MgCl2, 2.5 mM dNTP, 10 µM forward and reverse primers, 10% dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO, USA), and 5 units/µl of Takara Ex Taq™ (Takara Bio Inc., Shiga, Japan). The wsp PCR amplification was as follows: initial denaturation at 95℃ for 3 minutes, denaturation at 95℃ for 1 minute, annealing at 55℃ for 1 minute, an extension at 72℃ for 1 minute for 40 cycles, and a final extension at 72℃ for 3 minutes. The 16S amplification followed these conditions: initial denaturation at 95℃ for 2 minutes, denaturation at 95℃ for 2 minutes, annealing at 60℃ for 1 minute, an extension at 72℃ for 1 minute for two cycles, another 35 cycles of denaturation at 95℃ for 30 seconds, annealing at 60℃ for 1 minute, an extension at 72℃ for 45 seconds, and a final extension at 72℃ for 10 minutes. We included a positive control of a Wolbachia-positive Culex sp. and negative control of water in each PCR run.
PCR products were analyzed in 1.5% agarose gel stained with Midori Green Advance DNA stain at 100 V for 30 mins. To validate the presence of Wolbachia in each sample, we performed PCR amplification twice per marker. The criteria for a positive Wolbachia test were based on two successful amplifications per molecular marker, wsp, and 16S.
We also amplified the cox1 mitochondrial gene of Wolbachia-positive samples using the primer pairs LCO-1490 (5′-GGTCAACAAATCATAAAGATATTGG-3′) and HCO1-2198 (5′-AAACTTCAGGGTGACCAAAAAATCA-3′) [26]. We used the following PCR amplification profile: initial denaturation at 95℃ for 5 minutes, denaturation at 95℃ for 30 seconds, annealing at 48℃ for 45 seconds for 35 cycles, and a final extension at 72℃ for 7 minutes. The amplified PCR products were purified using QIAquick (Qiagen, Hilden, Germany) PCR Purification kits and sequenced by Eurofin Genomics Inc. Tokyo, Japan.
Identification of Wolbachia strains, haplotypes, and phylogenetic analysis
We assembled the forward and reverse sequences for each marker using the CodonCode Aligner version 1.2.4 software (https://www.codoncode.com/aligner/). We aligned the sequences using the online program MAFFT version 7 with the default settings (https://mafft.cbrc.jp/alignment/software/). We checked all generated sequences d for similarities with reference sequences from GenBank [27] using Basic Local Alignment Search Tool–Nucleotide BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch). Finally, we checked the sequence quality of the aligned sequences in Mesquite version 3.5 [28] by confirming the absence of stop codons.
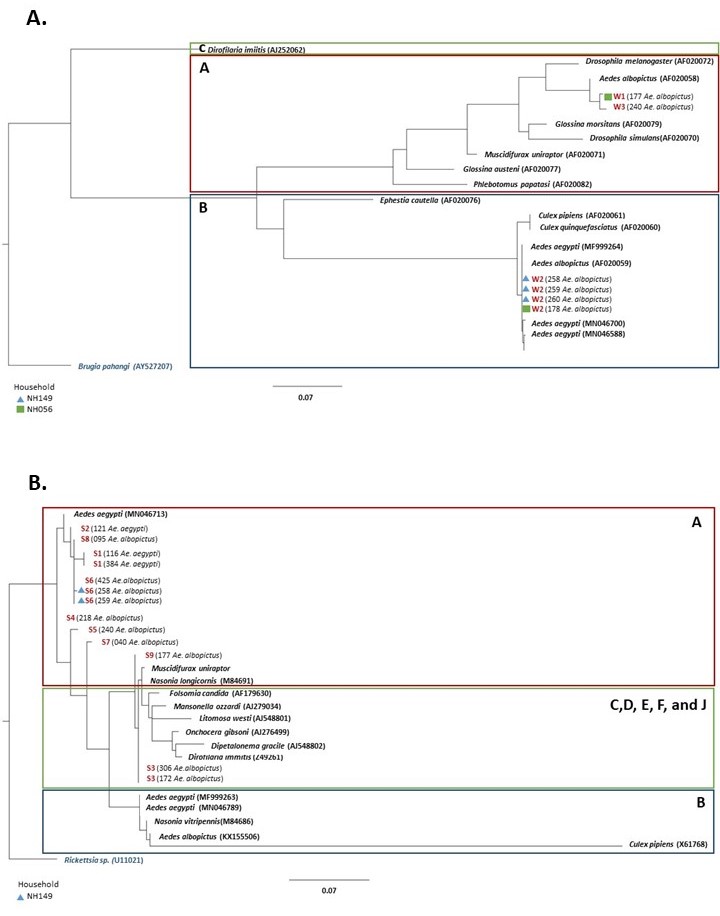
All wsp, 16S, and cox1 sequences were separately analyzed using DNAsp version 6.12.03 [29] to determine the number of haplotypes. We assessed the relationship of the Wolbachia strains of our study with representative sequences from different insect hosts by constructing a phylogenetic tree for the wsp and 16s sequences using PhyML 3.1 [30] using the default settings. We applied the GTR + G model for wsp and the GTR + G + 1 model for 16S. The model per marker was selected using the SMART model selection method [31]. We used Brugia pahangi (AY527207) and Rickettsia sp. (U11021) as the outgroups for the wsp and 16S, respectively.
{kind=link}