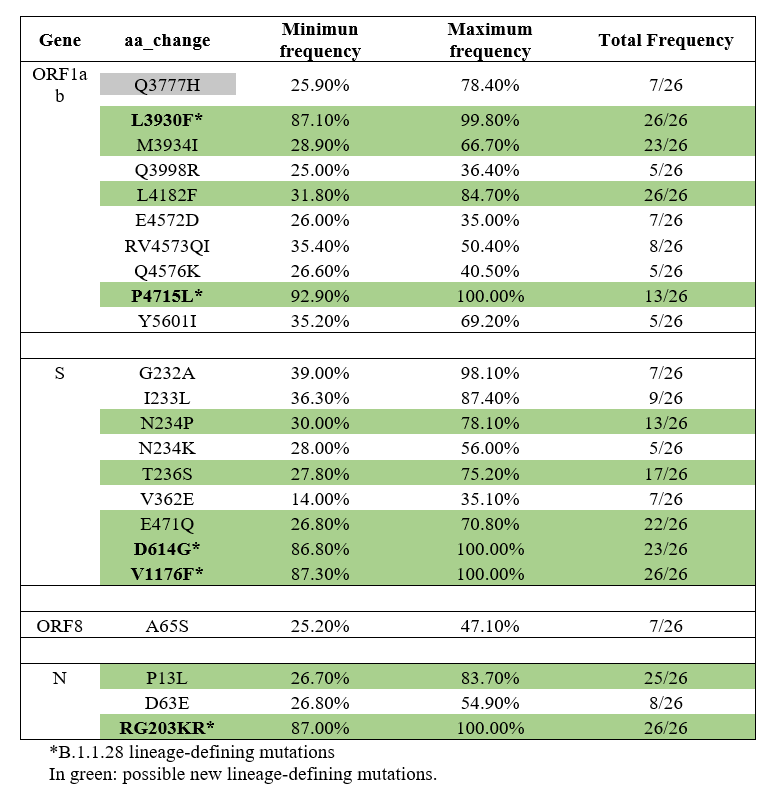
Consensus sequences were classified as B.1.1.28 according to the Pangolin COVID-19 Lineage Assigner tool (https://github.com/hCoV-2019/pangolin). Nevertheless, analyzing the FASTq reads through variant calling method, additional non-synonymous mutations were identified when compared to those of B.1.1.28. From 26 complete genomes, eight additional non-synonymous mutations were identified in ORF1ab (Q3777H, M3934I, Q3998R, L4182F, E4572D, RV4573QI, Q4576K, Y5601I), seven in S protein (G232A, I233L, N234P, N234K, T236S, V362E, E471Q), two in N protein (P13L, D63E) and one in ORF8 (A65S). Among those, six drew attention due to the total frequency in the samples and appear to be signatures of the putative new PX variant, specially N234P and E471Q in S protein, and M3934I and L4182F in ORF1ab (Table 1).
B.1.1.28 and P.2 were the most predominant strains in Brazil, until the introduction of P.1 [8], which replaced almost completely those two former variants. It’s important to highlight that B.1.1.28-defining lineage mutations were also found in ORF1ab (L3930F and P4715L), S protein (D614G and V1176F) and N protein (RG203KR), confirming the descent of this putative new lineage. It is likely that B.1.1.28 is evolving and that the findings described herein may represent a real-time scenario of this. The fact that the additional mutations have not yet been detected in all of the sequences is expected, as described in a P.1 introduction study, in which just 13% of B.1.1.28 sequences already presented E484K P.1 signature mutation [9]. Furthermore, it would not be surprising if the additional mutations are completely replaced in the coming months.
Among the likely P.X lineage S protein signatures, N234P was found in 13 genomes and E471Q in 23. Observing the presence of these mutations, the minimum and maximum frequency per sample was around 26-30% and 70-78%, respectively (Table 1). E471Q has already been detected in some countries, such as India, in a local circulating variants study [10], nevertheless, it has never been reported in Brazil, nor has it been associated with B.1.1.28 lineage. A systematic study based on all possible future S protein mutations tested E471Q and the results showed an increased binding affinity on the receptor-binding domain (RBD) [11], which can increase viral fitness.
ML phylogenetic tree showed that the sequences generated herein clearly clustered into a separated group in a highly supported monophyletic clade (Figure 1). The other clades were grouped and represent B.1.91, B.1.133, P.1, P.2 and B.1.1.8 lineages. Of all the aligned sequences, non-clustered with our sequences. The result reinforces the possibility of the new variant emergence, more related to B.1.1.28.
{kind=link}