Materials
Dulbecco’s modified Eagle’s medium (DMEM), minimum essential medium (MEM), foetal bovine serum (FBS), non-essential amino acids, Hanks balanced salt solution with calcium and magnesium (HBSS++), and phosphate-buffered saline (PBS) were from Gibco (Life Technologies Italia, Italy). Penicillin-streptomycin, L-glutamine, non-fat milk, bovine serum albumin (BSA), fatty-acid-free (faf)-BSA, Tween-20, MEM Eagle alpha-modified (α-MEM), Hoechst, and L-α-lysophosphatidylinositol sodium salt from soybean were from Sigma-Aldrich (St. Louis, MO, USA). Purified synthetic 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphoinositol (16:0 LPI), 1-stearoyl-2-hydroxy-sn-glycero-3-phosphoinositol (18:0 LPI) 1-oleoyl-2-hydroxy-sn-glycero-3-phospho-(1'-myo-inositol) (18:1 LPI), 1-arachidonoyl-2-hydroxy-sn-glycero-3-phosphoinositol (20:4 LPI) were from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). ML-191 and O1918 were from Cayman Chemical (Ann Arbor, MI, USA). Cannabidiol (CBD) was from Tocris Bioscience (Bristol, UK). Mowiol 4–88 and puromycin were from Calbiochem (San Diego, CA, USA). Lipofectamine 2000, Lipofectamine LTX with Plus reagent, Alexa488-tagged anti-mouse antibody, and Alexa546-labelled phalloidin, were from Invitrogen (Carlsbad, CA, USA). The mouse monoclonal anti-HA (16B12) antibody was from Covance (Princeton, NJ, USA). Paraformaldehyde was from Electron Microscopy Sciences (Hatfield, PA, USA). ‘Receptor-activator of nuclear factor kappa-Β ligand’ (RANKL) was from Peprotech (London, UK). Ionomycin, was from Santa Cruz Biotechnology (San Diego, CA, USA). All of the synthetic peptides were from Caslo ApS (Lyngby, Denmark). Based on the sequence of peptide-P1 (CKKNSPTLC), both a scrambled peptide (Scr; KCLTSNCPK) with the same amino-acid composition as peptide-P1 but a different primary sequence, and an irrelevant peptide (Irr_P; CGGNGPGLC) that included mutations to all of the polar amino acids of peptide-P1, were designed. All of the peptides were cyclised using an intramolecular disulphide bond between the two cysteine residues [38]. The fluorescent peptides were obtained by conjugation at the N-terminus with fluorescein-isothiocyanate (FITC) with an aminohexanoic acid linker. All other reagents were obtained at the highest purities available from Merck Life Science (Milano, Italy).
Site-directed mutagenesis
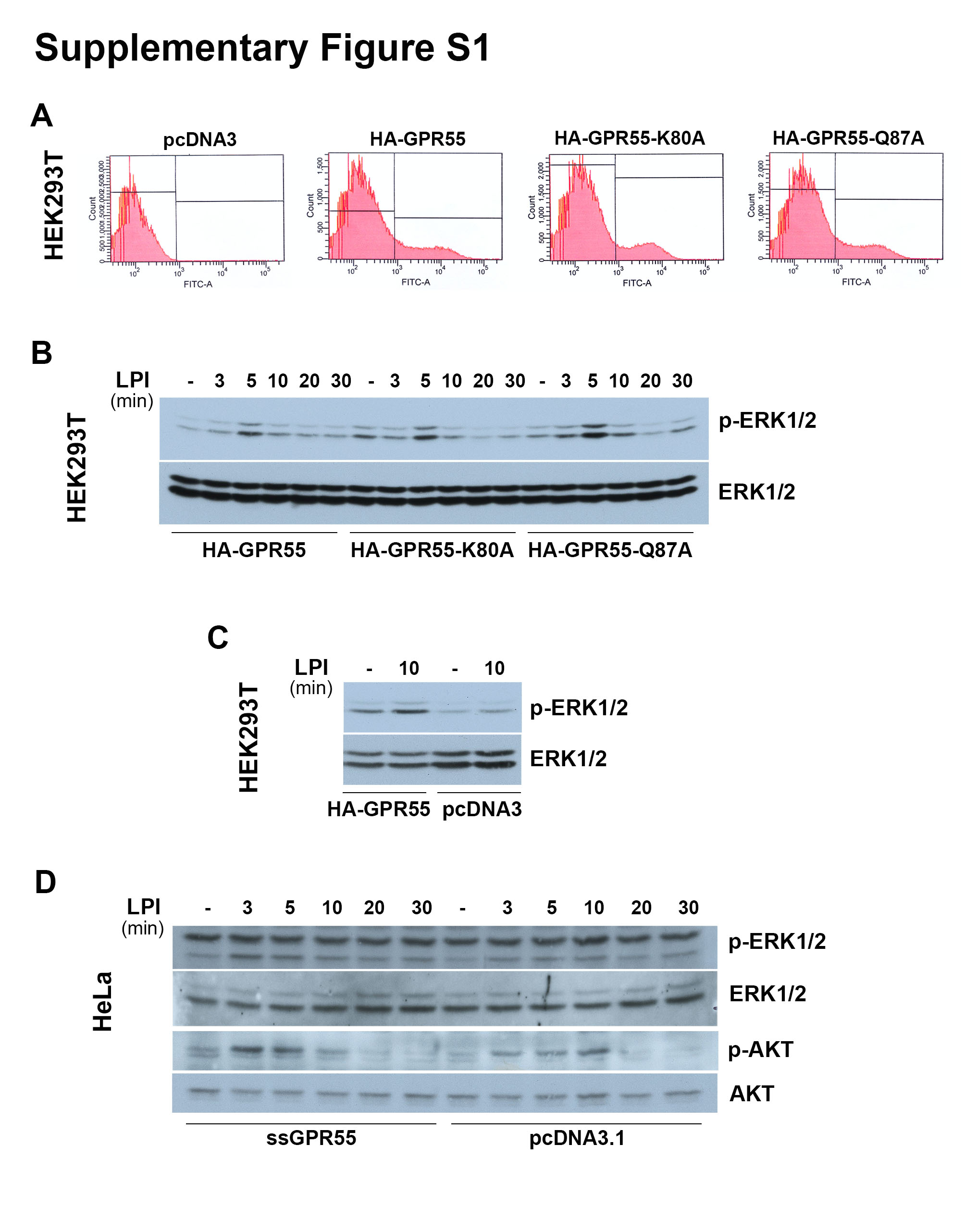
The construct of haemagglutinin (HA)-tagged human GPR55 in pcDNA3 (HA-GPR55) was a gift from Prof. K. Mackie, Indiana University, Bloomington, IN, USA [39], while the construct ss-3 × HA-GPR55 in pcDNA3.1 (ssGPR55) with a triple HA tag at the N-terminus and an optimised signal sequence (ss, derived from amino acids 1–33 of the human growth hormone: MATGSPTSLLLAFGLLCLPWLQEGSARDPPVAT) for efficient surface expression was from Prof. A. Irving, Dundee University, UK [40]. For both constructs, mutations were introduced by site-directed mutagenesis using QuickChange kits (Stratagene, La Jolla, CA, USA), according to the manufacturer instructions. The primers for the K80A mutation were 5’-CTCTCCCTCCCATTCGCGATGGTCCTGTCCCAG-3’ and 5’-CTGGGACAGGACCATCGCGAATGGGAGGGAGAG-3’ (Tm, 70.6 °C), and for Q87A were 5’-GTCCTGTCCCAGGTAGCGTCCCCCTTCCCGTCC-3’ and 5’-GGACGGGAAGGGGGACGCTACCTGGGACAGGAC-3’ (Tm, 73.1 °C).
RNA extraction and real-time PCR
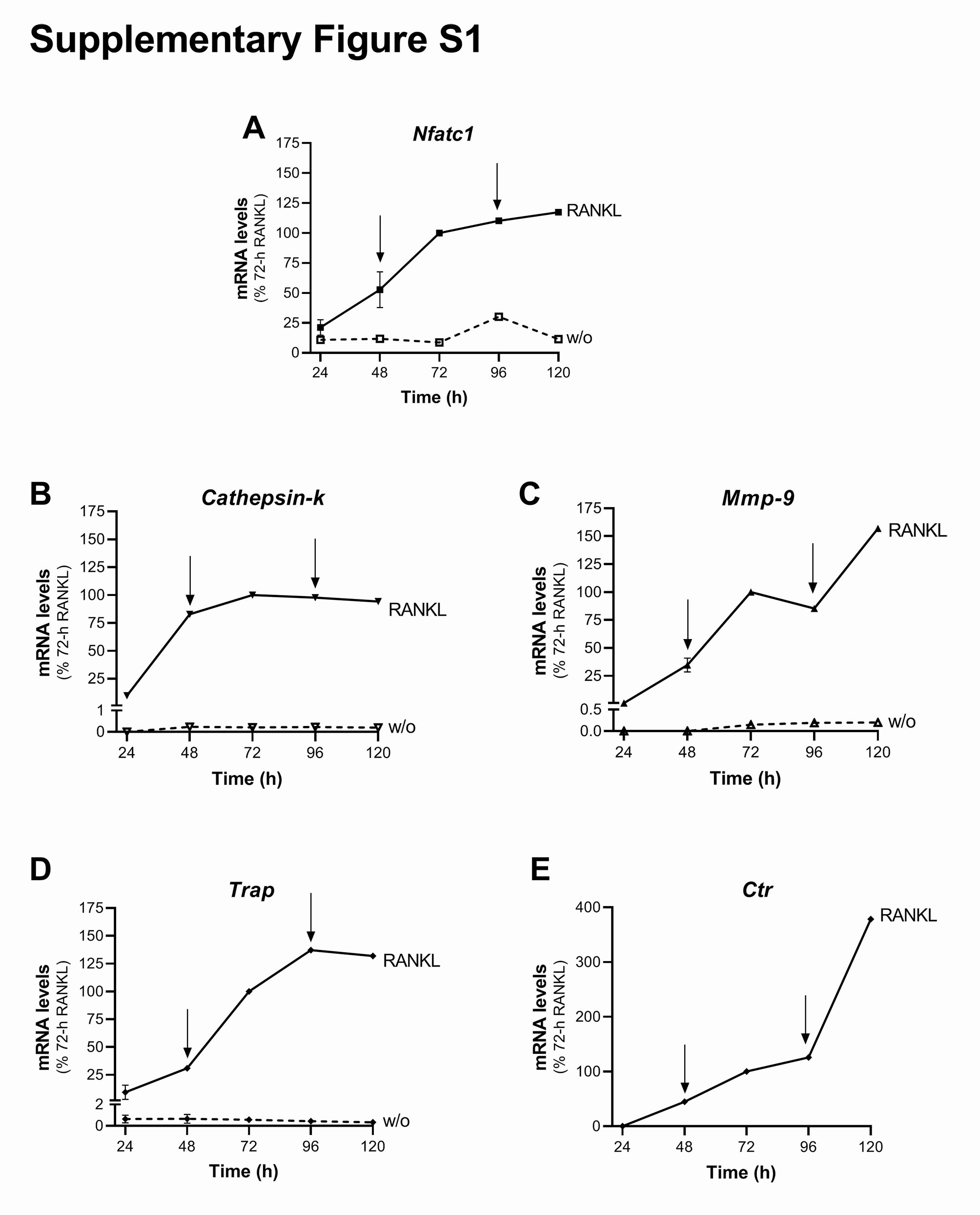
Total RNA was extracted using RNeasy isolation kits, cDNAs were obtained using QuantiTect Reverse Transcription kits, and real-time PCRs were performed with QuantiTect SYBR Green PCR kits (all from Qiagen, Hilden, Germany), according to the manufacturer instructions. The primers used for the real-time PCRs and their annealing temperatures are listed in the supplementary Table S1. Human hypoxanthine phosphoribosyltransferase 1 (HPRT1) or murine β2-microglobulin were followed as housekeeping genes. The real-time PCR programme consisted of an initial 15 min at 95 °C, and then 45 cycles as follows: 94 °C for 15 s, annealing temperature of each primer for 30 s, and 72 °C for 30 s. The real-time PCR machine used was a LightCycler 480 Instrument II (Roche, Indianapolis, IN, USA).
Cell culture
HEK293T cells were bought in 2012 from American Type Culture Collection (293T/17; ATCC catalogue number: CRL-11268), and were grown in monolayers in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin.
HeLa cells were received from Dr. Corda's laboratory (Institute of Biochemistry and Cell Biology, CNR of Naples) that bought them in 2006 from the European Collection of Cell Culture (ECACC catalogue number: 93021013). HeLa cells were maintained in MEM with 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin, and non-essential amino acids.
The RAW264.7 murine monocyte/macrophages were bought in 2003 from ATCC (catalogue number: TIB-71), and were cultured in DMEM with 10% heat-inactivated (30 min at 55 °C) FBS, 2 mM L-glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin.
All of the cells were tested free of mycoplasma, and were grown in a humidified atmosphere of 5% CO2 at 37 °C.
Transfection and RNA interference
For GPR55 overexpression, HEK293T cells were plated in their growth medium without antibiotics at 2.6 × 105 cells/well in 12-well plates, and 24 h later, the cells were transfected with 1 µg cDNA/well using Lipofectamine 2000, according to the manufacturer instructions. The pcDNA3 empty vector or that coding for human HA-GPR55 wild-type or its mutants HA-GPR55-K80A and HA-GPR55-Q87A were used.
HeLa cells and were plated at 1.5 × 105 cells/well in six-well plates in their growth medium without antibiotics. Twenty-four hours later, the cells were transfected with 2.5 µg/well pcDNA3.1, or the mutants ssGPR55-K80A, ssGPR55-Q87A, or 1.25 µg/well (complemented with 1.25 µg/well empty vector) ssGPR55, using Lipofectamine 2000, according to the manufacturer instructions. The different cDNA amounts were necessary to reach equivalent plasma-membrane expression of the receptors, as the mutants were expressed at lower levels, as verified by FACS analyses, and in line with previous reports [41].
For stable interference of GPR55, HeLa cells were plated at 1.5 × 105 cells/well in six-well plates in growth medium without antibiotics, and 24 h later, the cells were transfected with 1.7 µg/well OmicsLink short hairpin (sh)RNA expression clone CSHCTR001-CU6 (shCTRL) or clone HSH022476-3-CU6 (shGPR55) from GeneCopoeia (Rockville, MD, USA), using Lipofectamine 2000, according the manufacturer instructions. Forty-eight hours after transfection, HeLa clones stably expressing shRNAs were selected in growth medium containing 0.3 µg/mL puromycin. The efficiency of interference was monitored by real-time PCR using the primers listed in the supplementary Table S1. HPRT1 was followed as a housekeeping gene.
For transient interference of Gpr55, RAW64.7 cells were plated at 6 × 105 cells/well in six-well plates in growth medium without antibiotics. Twenty-four hours later, the cells were transfected with 250 pmol/well non-targeting small-interfering (si)RNAs (si-NT; siGENOME siRNA Pool #2; D-001206-14; Dharmacon, Chicago, IL, USA) or Gpr55-specific siRNAs (si-GPR55; siGENOME mouse GPR55 SMART pool; M-043590-01; Dharmacon) using Lipofectamine LTX and Plus Reagent, according to the manufacturer instructions. Twenty-four hours later, the cells were plated for the different assays or for RNA extraction. The efficiency of interference was monitored by real-time PCR after 72 h of interference, using the primers listed in the supplementary Table S1. β2-microglobulin was followed as a housekeeping gene.
Cell stimulation
Twenty-four hours after transfection (HEK293T, HeLa cells or clones) or 72 h after interference (RAW264.7 cells), the cells were washed twice with HBSS++, serum deprived (HEK293T cells for 4 h in DMEM; HeLa cells and clones for 2 h in MEM plus 2 mM glutamine and 25 mM HEPES; RAW264.7 cells for 2 h in DMEM), washed once again with HBSS++, incubated in stimulation buffer (HBSS++ with 10 mM HEPES, 0.4% faf-BSA for HEK293T cells; HBSS++ with 25 mM HEPES and 0.01% faf-BSA for HeLa cells, clones, and RAW264.7 cells) in the absence or presence of stimuli, at 37 °C for the indicated times. Incubations were terminated by washing the cells twice with cold HBSS++, and the analyses were performed as reported below.
Western blotting
Cell lysates were obtained by scraping the cells into phospho-lysis buffer: 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 5 mM EDTA, 1% Triton X-100, 50 mM NaF, 40 mM β-glycerophosphate, 200 µM sodium orthovanadate, plus protease and phosphatase inhibitors (Roche). Following gentle homogenisation by 20 passages through a 26-gauge needle, the lysates were centrifuged at 10,000 × g for 5 min at 4 °C, and the supernatants were collected.
Protein lysates were subjected to SDS-PAGE, and after electrophoresis, the proteins were transferred to a nitrocellulose membrane (PerkinElmer Life Science, Boston, MA, USA). For immunoblotting, the membranes were blocked with 5% non-fat milk in TBS (10 mM Tris-HCl, pH 7.4, 10 mM NaCl) plus 0.1% Tween-20 (T-TBS) for 30 min at room temperature, and incubated with primary antibodies in T-TBS plus 3% BSA for 2 h at room temperature, or overnight at 4 °C. The membranes were washed twice in T-TBS for 7 min, and then incubated with secondary antibodies conjugated to horseradish peroxidase (1:5,000) (Calbiochem, San Diego, CA, USA) in T-TBS with 5% non-fat milk for 30 min at room temperature. The membranes were then washed twice with T-TBS and once with TBS for 5 min, and the signals were detected by ECL (Amersham Pharmacia, Piscataway, NJ, USA). The rabbit anti-phospho AKT (Ser473), anti-phospho p38 (Thr180/Tyr182), anti-phospho p42/44 (Thr202/Tyr204), anti-p38 (all at dilution 1:1000) were from Cell Signaling Technology (Danvers, MA, USA). The rabbit anti-AKT (B-1), and anti-p42/44 (ERK1; K-23) were from Santa Cruz Biotechnology.
Ca2+ assay
After 48 h of siRNA treatments, the RAW264.7 cells were detached with 600 µM EDTA in PBS, and plated at a density of 8 × 104 cells/well in 96-well plates. Seventy-two hours from the interference, the cells underwent Ca2+ measurements using Fluo4-NW Calcium Assay kits (Invitrogen), according to the manufacturer instructions. Interfered cells were washed twice with HBSS++, incubated with 50 µL loading buffer (0.01% faf-BSA, 20 mM HEPES in HBSS++, 5 mM probenecid, and 2 × Fluo4-NW) for 45 min at 37 °C. All the subsequent incubation steps were performed at 37 °C within the Fluoroskan Ascent FL (Thermo Fisher Scientific, Waltham, MA USA) and the fluorescence recorded with an Ex 485/ Em 520 every 3 s. The baseline fluorescence was monitored for 5 min, then 50 µL assay buffer (0.01% faf-BSA, 20 mM HEPES in HBSS++) was added without or with 10 µM 16:0 LPI, and fluorescence was recorded for a further 5 min. Subsequently, the cells were stimulated with addition of 2 µL ionomycin (1 µM final concentration, for Fmax), 2 µL EGTA (6 mM final concentration, Fmin) and 2 µL CaCl2 (8 mM final concentration) in sequence, and the fluorescence recorded for 2 min for each stimulus. The intracellular Ca2+ concentrations were calculated according to Eq. (1):
[Ca2+]free = Kd [F-Fmin/[Fmax-F] (1),
using the Fluo-4 Kd of 345 nM.
Cytoskeleton analysis
Twenty-four hours after plating the RAW264.7 cells at a density of 1.2 × 106 cells/well in six-well plates, or 2.5 × 105 cells/well in 24-well plates on coverslips, the cells were serum deprived for 2 h and then stimulated while adhered, with LPI in the assay buffer (0.1% faf-BSA, 20 mM HEPES in HBSS++). Stimulation was blocked by two washes with HBSS++, and cells on coverslips were processed for immunofluorescence (see above), while the cells in the six-well plates were scraped into cytoskeleton buffer (10 mM 4-morpholineethanesulfonic acid, 150 mM NaCl, 5 mM EGTA, 5 mM MgCl2, 5 mM glucose), for FACS analysis. Then, an equal volume of fixation solution (1% Triton X-100, 0.5% glutaraldehyde, in cytoskeleton buffer) was added to the cell suspension and left for 2 min at room temperature. The cells were then washed twice (5 min each) with cytoskeleton buffer, fixed again for 15 min with 1% glutaraldehyde in cytoskeleton buffer at room temperature, further washed three times (10 min each) with cytoskeleton buffer, treated with 500 mg/mL sodium borohydride for 10 min on ice, and washed three times (10 min each) with cytoskeleton buffer. Finally, the fixed cells were stained with 33 nM Alexa546-labelled phalloidin for 1 h at room temperature, washed three times (10 min each) with cytoskeleton buffer, suspended in PBS with 3% BSA, and analysed by FACS (FACSCalibur or FACSAria III; Becton Dickinson, Franklin Lakes, NJ).
Immunofluorescence microscopy
For actin staining, the cells were rinsed with HBSS++, fixed in 4% (v/v) paraformaldehyde for 10 min at room temperature, and permeabilised with blocking solution (50 mM ammonium chloride, 0.5% BSA, 0.1% saponin, 0.02% NaN3, in PBS), for 30 min at room temperature. The cells were stained for a further 1 h at room temperature with 33 nM Alexa488-labelled phalloidin for filamentous actin visualisation, and 2 µg/mL Hoechst for nucleus staining, with all of the reagents diluted in blocking solution. Then the cells were washed three times with PBS plus 0.02% Tween-20, and the coverslips were mounted with Mowiol 4–88 and examined under confocal microscopy (LSM 510; Zeiss, Oberkochen, Germany). The nuclei of multinucleated cells were counted in a blinded manner using a 63 × objective, moving across the coverslip along the vertical and horizontal directions.
GPR55 quantification by FACS
Twenty-four hours after transfection, the HEK293T cells were detached with PBS plus 1 mM EDTA, centrifuged at 300 × g for 5 min at 4 °C, incubated in blocking buffer (5% BSA, 5% FCS, in PBS) for 30 min on ice, and then centrifuged at 300 × g for 5 min at 4 °C. All of the subsequent steps were on ice with cold PBS plus 3% BSA. The cells were stained with a murine anti-HA antibody (1:1,000) for 1 h, washed three times, further incubated in the dark with an Alexa488-tagged anti-mouse antibody (1:800) for 30 min, washed three times, suspended in PBS plus 3% BSA, and analysed by FACS.
For the GPR55-internalisation assay, after stimulation, the HeLa cells were washed twice with cold HBSS++, stained while adhered with the monoclonal anti-HA antibody (1:1,000) in PBS plus 3% BSA for 1 h on ice, washed three times with cold PBS, incubated with the Alexa488-tagged anti-mouse antibody (1:800) in PBS plus 3% BSA for 45 min on ice. After two washes with cold PBS and a final wash with PBS at room temperature, the cells were detached by scraping. The collected cells were centrifuged at 300 × g, suspended in PBS plus 3% BSA, and analysed by FACS.
Peptide binding to RAW264.7 cells
Wild-type or 48-h-interfered RAW264.7 cells were plated at a density of 1.2 × 106 cells/well in six-well plates, and the following day they were used for on-plate-binding assays, or for RNA extraction. Before peptide addition, the cells were washed twice with HBSS++, and then incubated without or with 40 µg/mL FITC-P1 or FITC-Scr for the indicated times at 37 °C in HBSS++ plus 0.01% faf-BSA. The incubations were stopped by three washes with PBS, detached by scraping with PBS plus 2 mM EDTA, and then suspended in PBS plus 3% BSA. Fluorescence intensity was evaluated by FACS, and reported as means of cell-associated fluorescence increases compared to cells incubated in absence of peptides.
To evaluate peptide-P1 specificity towards murine GPR55, RAW264.7 cells were Gpr55-interfered as above, but the duplexes were previously mixed with the double pmol amount of siGLO Red transfection indicator (Dharmacon). Under these conditions, a 40% reduction in Gpr55 mRNA levels was measured in the total population of siGPR55-treated versus siNT-treated cells, and 30% of both populations were siGLO-positive, as verified by FACS analysis. Peptide binding was evaluated only towards siGLO-positive cells, which were assumed to have a higher proportion of siRNA-treated cells compared to the total population. FITC-fluorescence intensity was evaluated by FACS, and reported as means of cell-associated fluorescence increases compared to cells incubated in absence of peptides.
Osteoclastogenesis in-vitro assay
For the osteoclastogenesis in-vitro assay, RAW264.7 cells were plated in differentiation medium (α-MEM with 10% heat-inactivated FBS, 2 mM L-glutamine, 100 U/mL penicillin, and 100 ug/mL streptomycin) at a density of 5 × 103 cells/well in 24-well plates on coverslips for morphological analysis, or at 2 × 104 cells/well in six-well plates for RNA extraction. Twenty-four hours later and every 48 h, the medium was replaced and the cells were treated with 15–30 ng/mL RANKL with DMSO and/or PBS as carriers, or with GPR55 antagonists/ agonist (0.5 µM ML-191, 30 µM O1918, 0.5 µM CBD, 1 µM soybean LPI), or with the peptides (150 nM peptide-P1; or the irrelevant peptide, Irr_P). Twenty-four hours after the last addition, the cells were fixed with 4% paraformaldehyde for morphological analyses or harvested in lysis buffers for RNA extraction (see above).
Statistical analysis
Statistical analysis was performed with the GraphPad Prism software (GraphPad Software, Inc. La Jolla, CA, USA). Comparisons between groups were performed using Student’s t-test and Analysis of Variance (ANOVA) with 95% confidence interval. p < 0.05 was considered statistically significant.
{kind=link}
{kind=link}