Patient Characteristics
The characteristics of the subjects in each group are shown in Table 1. All eight participants within the ASP group were female, and the median age was 55 years (range 49, 31–80 years). Five females and five males were assigned to the ATLL group, and the median age of the entire group was 41 years (range 49, 24–73 years). In the leukemic group, six had acute ATLL, three had chronic, and one had lymphomatous types of ATLL. HTLV-1 proviral load levels varied from one copy to 208 copies/103 PBMCs in the ASP group and varied from 104 to 4482 copies/103 PBMCs in the ATLL group (two-tailed p-value = 0.02).
Description of whole-genome sRNA-sequencing data from each group
sRNA sequencing generated a total of 149,424,024 reads by Illumina MiSeq platform. After the exclusion of low quality reads, 124,171,839 clean reads were mapped to selected genic regions. The filtered sRNA reads for each sample were aligned to the human genome sequence dataset are depicted in Table 1. The MPS approach yielded 381 sRNA molecules consisting of 307 and 74 known and novel sRNAs, respectively. Of the 307 known sRNA, 203 were 52 tRNA miRNA, 25 snoRNA, 12 scRNA, 5 scRNA_pseudogene, 7 snRNA, and 3 piRNA (Table S1). The analysis also identified 234 mature miRNA, with 22 had expression greater than a two-fold change. Of the 74 novel genes, 8 were novel miRNA, 18 snoRNA, and 48 unknown (Table S2).
Expression profiling of the known sRNAs
Among the 307 known sRNAs identified in this study, 226 sRNAs were differentially expressed between the three groups (p (corr) < 0.05), 184 and 26 were up and down-regulated, respectively, in both ATLL and ASP group than in HC group (Table S3). The expression profiles of the 226 above-identified sRNA were included in the subsequent analysis. Unsupervised cluster analysis based on the Euclidean distance was used to visualize their expression profiles. The results revealed a differential pattern of expression with five main clusters in which acute type-ATLL, other clinical type-ATLL, ASP, and HC group were segregated (Fig. 1). Interestingly, one patient (005ATLL) classified as chronic-type ATLL at the time of sample collection, progressed to aggressive acute phase after two years and died with leukemia five months after the onset of the acute phase. Another observation of this analysis is that the sRNA profiles of patients with nonacute ATLL- type in some instances even more similar to the ASP group. The first cluster is constituted of 61 sRNAs with expression profiles of lower expression levels in the HC group. In the second cluster, a differential expression pattern was observed with 21 upregulated genes in HC and ATLL patients. The 3rd, 4th, and 5th clusters revealed 27, 19 (mostly snoRNA), and 98 genes, respectively, with an expression pattern of positive regulation in ASP and nonacute ATLL –type patients.
In further analysis, we aimed to identify differentially known and novel expressed sRNAs using one-way ANOVA and pairwise contrasts, with the Benjamini-Hochberg[20] correction (p < 0.05) on the samples in the three groups. In the ATLL group, we found different expression levels of 41 of the known sRNAs from the ASP and HC groups, of which 15 and 26 were up-and down-regulated sRNAs, respectively (Table S3). Next, after correction for multiple testing (Benjamini-Hochberg) and application of Moderated t-Test p cutoff = 0.05, we compared the expression profiles of the known sRNA between the ATLL group and HC subjects. The results indicated that 114 of the 226 known sRNA remained significantly differentially expressed, of which 100 were upregulated and 14 were downregulated. The upregulated sRNA in the ATLL group includes 70 miRNA, two piRNA, 8 snoRNA, 7 snRNA, and 13 tRNA. On the other hand, the down-regulated entities include 13 miRNA and only one scRNA_pseudogene. hsa-mir-150, trna65, has-let-7b, and hsa-mir-331 were of the highly expressed sRNAs (p (corr) < 0.05) (Table S4). The most four strongly downregulated sRNAs in PBMCs of the ATLL group were hsa-mir-19a, hsa-mir-19b-1, hsa-mir-19b-2, hsa-mir-7-3, and hsa-mir-7-2, among which, the hsa-mir-19a had greater than − 27 of ranking fold changes.
Then, we used the same strategy to compare the expression profiles of the known sRNA of the ASP vs. HC group. The analysis revealed 216 of the 226 known genes satisfying corrected p-value cutoff < 0.05, and most of them (90.3%) were upregulated in the ASP subjects (Table S5). The 21 downregulated sRNAs includes 20 miRNA and only one tRNA, namely trna9. The upregulated entities in the ASP group include 121 miRNA, 3 piRNA, 9 scRNA, two scRNA_pseudogene, 23 snoRNA, 7 snRNA, and 30 tRNA. The most strongly upregulated five sRNAs in ASP group were hsa-mir-150, hsa-mir-146a, hsa-let-7e, hsa-mir-342, and hsa-mir-28. The most five significantly downregulated sRNAs in PBMCs of the ASP group were hsa-mir-486, hsa-mir-486-2, hsa-mir-183, hsa-mir-144, and 451a.
Finally, we evaluated the differentially expressed genes between ATLL and ASP. The data showed that of the 226 known sRNAs, 149 were remained differently expressed and that 140 of them were downregulated (Table S6). All of the nine-upregulated genes in the ATLL group were miRNAs, of which hsa-mir-451a and hsa-mir-183 were the top two-upregulated entities. The 140 down-regulated sRNA include 100 miRNA, one piRNA, 8 scRNA, two scRNA_pseudogene, 21 snoRNA, 7 snRNA, and 17 tRNA. The most five strongly downregulated sRNAs in PBMCs of the ATLL group compared to ASP subjects (p (corr) < 0.05) were hsa-mir-26a-2, hsa-mir-26a-1, trna15, hsa-mir-4772. Among these, hsa-mir-26a-2 was the most downregulated sRNA in both Benjamini–Hochberg FDR (0.001) and ranking of fold change − 10.
A Venn diagram representation of the number of all sRNAs overlapping in different comparisons among the three groups is depicted in Fig. 2. The results showed 60 sRNAs were common among the 3 groups, while 5 sRNAs, 15 sRNAs, and 3 sRNAs were specific in entity 1 (ATLL vs. HC), entity 2 (ASP vs. HC), and entity 3 (ATLL vs. ASP), respectively.
Expression profiling of the novel sRNAs
Sequencing of the library and subsequent analysis of the three groups revealed 74 new sRNAs, of which 51 were differently expressed (28 unknown, 16 snoRNAs, and 7 miRNAs). After correction for multiple testing, the data showed that 26 of the 51 novel sRNAs remained significantly dysregulated in the ATLL vs. HC group; 17 and 9 sRNAs were up- and down-regulated, respectively. The reads of these 26 novel genes were annotated as 14 unknown novel genes, 3 miRNAs, and 8 snoRNA (Table S7). Comparison of data after correction for multiple testing between the ATLL and ASP demonstrated that 27 novel sRNAs were strongly dysregulated; 6 and 21 sRNAs were up- and down-regulated, respectively. The 27 new sRNA include 14 unknown novel genes, 9 snoRNA, and 4 miRNA (Table S8). All of the four novel sRNA that displayed high expression abundance were unknown sRNAs. Of the 5 top strongly down-regulated sRNA, three were unknown and one was miRNA and snoRNA each. Finally, the same strategy of analysis was used to compare the ASP and HC group. The results revealed significant dysregulation of 48 genes in the ASP subjects when compared to the HC group and were annotated as 27 unknown novel genes, 6 miRNAs, and 15 snoRNA (data no shown). Among these significantly deregulated novel genes, 34 and 14 sRNAs displayed high and low expression abundance, respectively. The four top strongly upregulated novel sRNAs were annotated as two miRNA and snoRNA each, whereas 100% of the novel entities that displayed low expression abundance were annotated as unknown genes.
Expression profiling of the known mature miRNAs
To reduce a bias depending on the coverage, we considered only known mature miRNAs covered by at least 20 reads. A total of 234 mature miRNAs fulfilled the criteria, 164 of which were significantly differentially expressed in PBMCs among the 3 groups. For known mature miRNAs after correction for multiple testing, 49 of the 164 active miRNAs remained significantly dysregulated (p (corr) < 0.001) (Table S9). The top 10 most highly expressed miRNAs among all samples were hsa-miR-199a-3p, hsa-miR-26a-5p, hsa-miR-199b-3p, hsa-miR-150-5p, hsa-let-7d-3p, hsa-miR-155-5p, hsa-miR-26b-5p, hsa-miR-222-3p, hsa-miR-181b-5p, and hsa-miR-30e-3p. The individual analysis between the ATLL and HC subjects displayed nine entities of 55 satisfying the corrected p-value (Benjamin-Hochberg FDR) cutoff < 0.001; eight and one miRNAs were up- and down-regulated, respectively (Table S10). Among the nine miRNAs, the uniquely down-regulated hsa-mir-19a-3p and the upregulated hsa-let-7d-3p followed by 199b-3p, miR-331-3p, and hsa-mir-199a-3p exhibited the most prominent differences. In the analysis of miRNAs of ATLL vs. ASP, three of the 55 miRNAs, hsa-mir-146b-5p, hsa-mir-26a-5p (Entrez 407015), and hsa-mir-26a-5p (Entrez 407016) showed considerably low expression levels (p (corr) < 0.001). Finally, the differential regulation of miRNA between the ASP and HC were computed and revealed 48 of the 49 active miRNAs displaying high (n = 46) and low (n = 2) expression abundance. hsa-mir-150-5p, hsa-mir-146a-5p, hsa-mir-342a-3p, hsa-mir-181a-2-3p, and hsa-mir- 23a-3p were annotated as the five top considerably upregulated genes. hsa-mir-451a and hsa-mir-19a-3p were the unique miRNA that displayed low expression abundance (Table S11).
We then used the mirWalk algorithm to generate a network and hence determine and rank the miRNA-mRNA putative target interaction. As shown in the interaction map (Fig. 3), the blue circles are ATLL-relevant mRNAs targeted individually by, for example, miR-155-5p, miR-150-5p, miR-30c-5p, and miR-98-5p. ATLL-specific mRNAs were targeted, for instance, by miR-26a-5p, miR-26b-5p, miR-331-3p, and miR-378a-3p.
Validation by qRT-PCR Assay
To further validate the screen of miRNA expression with an independent qRT-PCR method using the samples from a total of nine ATLL patients used for sequencing, we chose two of the most differentially regulated miRNAs, hsa-mir-451a and hsa-mir-144, among ATLL-relevant miRNAs. As shown in Fig. 4, the expression levels of the selected mir-451a were significantly downregulated in ATLL patients compared with the ASP group. The level of hsa-mir-144 could not be accurately quantified in most samples because of its low abundance or absence.
Target genes analysis
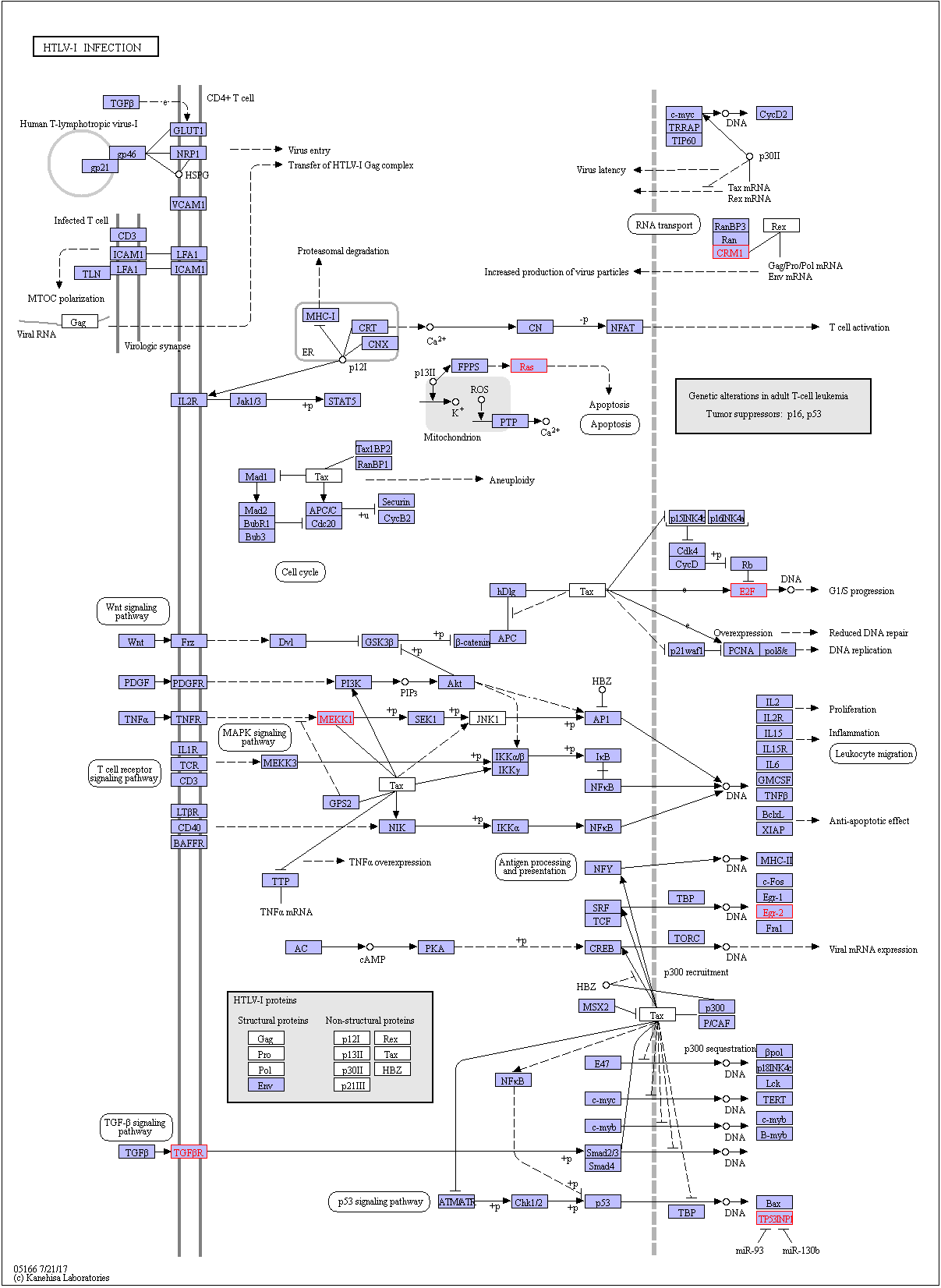
By applying increasingly stringent statistical methods, the current study has identified 49 non-redundant mature miRNAs that were significantly dysregulated in PBMCs of HC and ASP groups compared with ATLL patients (p (corr) < 0.001). Because miRNAs regulate target genes expression at the post-transcriptional level, we predicted the target genes of dysregulated miRNAs using the miRWalk 3.0 algorithm, which provides a comprehensive list of putative and validated mRNAs for a given miRNA by combining results from 12 established target prediction algorithms for the identification of putative miRNA targets. A total of 404 non-redundant genes were predicted as putative target genes of dysregulated miRNAs (Table S12). The total numbers of high confidence mRNA hits generated by hsa-let-7b-5p, hsa-let-7a-5p, hsa-miR-130b-3p, hsa-let-7d-5p, and hsa-miR-27b-3p were 62, 53, 34, 33, and 51, respectively. Our manual approach for the identification of ATLL-relevant mRNAs targeted by the selected miRNAs yielded in mRNAs XPO1, TGFBR1, EGR2, NRAS, SMAD2, PIK3R1, E2F2, TP53INP1, and MAP3K1 (supplementary Fig. 1).
GO/pathway enrichment analysis of target genes
To understand the role of miRNAs in the progression of ATLL in HTLV-1 infected patients, GO and KEGG pathway enrichment analysis were carried out. The top GO terms at the MF level were; cytokine-mediated signaling pathways and transcription initiation from RNA polymerase II promoter. The top GO terms at the CC level were; nuclear chromatin and promyelocytic leukemia body. The top GO terms at the MF level were; RNA polymerase II proximal promoter sequence-specific DNA binding and transcription_factor_binding. GO analysis results showed that upregulated DEGs were significantly enriched in binding, protein binding, and organic cyclic compound binding at MF level. The KEGG analysis revealed that the upregulated differentially expressed miRNAs were mostly enriched in various pathways, including Human T-cell leukemia virus 1 infection and cancer as the most significantly enriched pathways (FDR corrected p-value of ≤ 0.05). Of note, pathways in cancer is a combination of several pathways, including pathways reported to play a key functional role in ATLL pathology (TGF-β, Wnt, p53, apoptosis, and MAPK signaling), indicating that several vital processes might be regulated by these miRNAs (supplementary Fig. 2).
{kind=link}