Microbiome diversity and community structure in vanilla. Our metabarcoding data revealed that a total of 1,054,997 16S reads passed quality filtering and clustered into 1666 bacterial OTUs. The most species-rich phyla were Proteobacteria (573 OTUs), Actinobacteria (277), Bacteroidetes (245), Acidobacteria (125) and Planctomycetes (124), while 97 OTUs remained unassigned at phylum level. OTU richness was significantly higher in root (1594 OTUs) compared to stem (693) samples when comparing both observed richness (χ2 = 9.9265, p-value = 0.002) and Shannon index (χ2 = 8.6471, p-value = 0.003). When looking at species diversity per condition (Table 1), root OTU richness was higher in symptomatic (1385 OTUs) and asymptomatic (1321) compared to feral (763) individuals, while stem OTU richness was higher in feral (598) compared to symptomatic (305) and asymptomatic (163) individuals. These differences however remain non-significant based on pairwise Wilcoxon tests for both observed richness and Shannon index, potentially due to the low number of samples studied. Based on NMDS ordination (Figure 1), the bacterial community composition varied most significantly between organs (R2 = 0.91, p-value = 0.001), while the effect of condition (R2 = 0.03, p-value = 0.003) and the combination of both factors (R2 = 0.06, p-value = 0.002) were lower but nevertheless significant.
Regarding fungi, 1,117,927 ITS2 reads passed quality-filtering and clustered into 889 fungal OTUs belonging to the phyla Ascomycota (607 OTUs), Basidiomycota (121), Mucoromycota (12, sensu Spatafora et al. 2016), Rozellomycota (6), Chytridiomycota (1) and Kickxellomycota (1), while 141 OTUs remained unassigned at phylum level. In addition, 403 OTUs remained unassigned at family level, emphasizing the large number of undescribed fungal groups that are associated with vanilla. The most species-rich families were Nectriaceae (32 OTUs), Herpotrichiellaceae (22), Aspergillaceae (14), Cyphellophoraceae (10) and Glomerellaceae (6). Similarly, to what was observed in the bacteria dataset, OTU richness was significantly higher in root (772 OTUs) than in stem (363) samples when comparing observed richness (χ2 = 8.04, p-value = 0.005), but not Shannon index (χ2 = 3.19, p-value = 0.07). When looking at species diversity per condition (Table 1), root OTU richness was higher in symptomatic (510 OTUs) than in asymptomatic (438) and wild (303) individuals, while stem OTU richness was higher in feral (226) compared to symptomatic (112) and asymptomatic (98) individuals. Similarly, to what was observed in the bacteria dataset, these differences remain non-significant based on pairwise Wilcoxon tests for both observed richness and Shannon index, potentially due to the low number of samples studied. Fungal community composition varied most significantly between organs (R2 = 0.66, p-value = 0.001), but a good portion of the variation was also explained by condition (R2 = 0.18, p-value = 0.001) and the combination of both factors (R2 = 0.11, p-value = 0.003).
Fungal diversity detected from culture isolates. Microbiological isolation of vanilla roots and stem led to 100 fungal isolates growing on PDA, 34 on Sabourad and 10 on Czapek media, for a total of 144 axenic cultures. The isolates were grouped into 58 morphotypes for which we obtained ITS sequences that were deposited in GenBank (MZ270645-MZ270702) (Table S1). Based on ITS sequences we were able to identify 14 genera, predominantly from the phylum Ascomycota, except for two isolates belonging to Basidiomycota. 41 out of 51 morphotypes were identified at family level, distributed in 8 families: Xylariaceae (18 morphotypes), Nectriaceae (7), Stachybotryaceae (5), Biatriosporaceae (4), Bionectriaceae (2), Irpiceae (2), Trichosphaeriaeae (2) and Aspergillaceae (1). In the Xylariaceae ITS phylogenetic tree, our sequences are grouped within the genera Daldinia, Xylaria and Entonaema (Suppl. Fig. S2). When comparing Xylariaceae ITS sequences in UNITE database, the 18 sequences clustered into only four SH identified as Hypoxylon griseobrunneum, Xylaria multiplex, Xylariaceae and Daldinia starbaeckii, each corresponded to an OTU from the metabarcoding analysis. These results indicate that Xylariaceae SH and OTUs may show a high phenotypic diversity of morphotypes when isolated in culture media; and it also suggests that the use of morphotypes is limited in identifying and selecting Xylariaceae. Interestingly, these 4 cultivable OTUs, were present only in asymptomatic samples according to metabarcoding analysis. The second most-species rich family recovered from our cultures was Nectriaceae that includes the genus Fusarium. Nectriaceae ITS sequences from vanilla clustered in the phylogenetic tree with Fusarium solani, Fusarium oxysporum and Fusarium incarnatum-equiseti species complexes (Suppl. Fig. S1). In the UNITE database, these Fusarium sequences were identified as SH corresponding only to Fusarium pseudensiforme and Gibberella fujikuroi. In addition, according to metabarcoding data, Nectriaceae morphotypes sequences matched with 6 different OTUs identified as Fusarium, Nectriaceae and Sordariomycetes in the metabarcoding analysis. The OTUs matching these isolates were present predominantly in the root samples (Table S1).
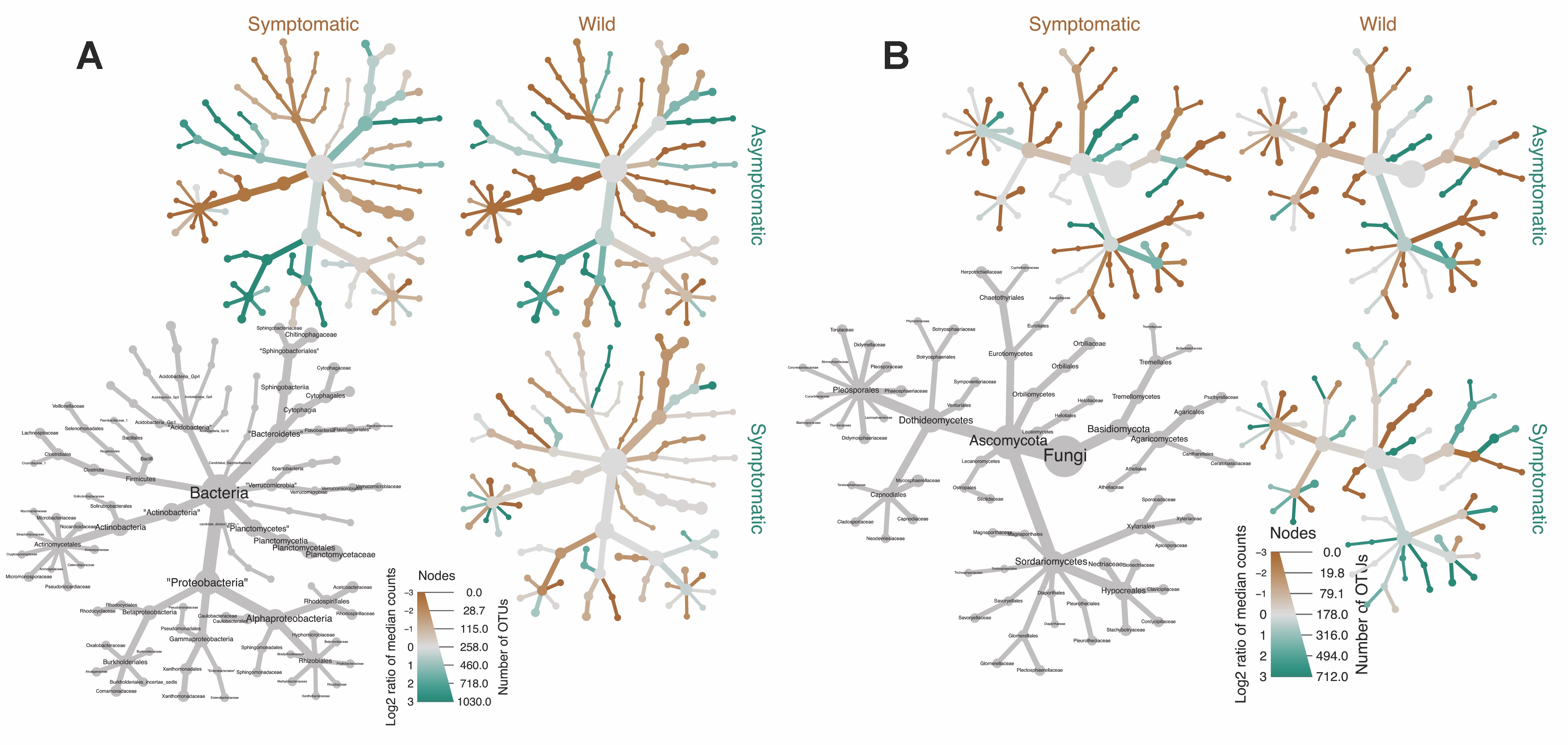
Most abundant taxa in vanilla microbiome. Even though the frequency of bacterial and fungal phyla is similar between conditions in both root and stem samples (Figure 2), when analyzing the 10 most frequent bacterial and fungal families in our dataset, marked differences in OTU frequencies were observed between both condition asymptomatic, cultivated/symptomatic, cultivated/feral (asymptomatic, symptomatic, and feral) and organ type. Unfortunately, because of the very small number of feral samples, we could not analyze differences between feral and cultivated (as two states). Asymptomatic stems shared the bacterial family Sphingomonadaceae, and the fungal families Phaeosphaeriaceae, Orbiliaceae and Glomerellaceae, while Pseudonocardiaceae (bacteria) and Nectriaceae (fungi) were almost absent. The relative abundance among bacterial taxa (Figure 3) shows that the family Beijerinckaceae as well as various Actinobacteria were significantly more abundant in feral individuals, while Proteobacteria were significantly more abundant in asymptomatic individuals. The class Negativicutes was also significantly more abundant in asymptomatic individuals than in symptomatic individuals. Commonly isolated bacterial genera included Paenibacillus from orchid meristems [38] and Streptomyces, Bacillus, Flavobacterium, and Pseudomonas from orchid roots [39,40], which we recovered in almost all our samples. Regarding fungi, the families Mycosphaerellaceae, Neodevriesiaceae Sporocadaceae, Stachybotryaceae, Teratosphaeriaceae, Tremellaceae, Xylariaceae, as well as the order Chaetothryriales were significantly more abundant in symptomatic individuals (Fig. 3). Mycosphaerellaceae was also significantly more abundant in feral than asymptomatic individuals, while Neodevriesiaceae was less abundant in feral plants than cultivated symptomatic individuals. Although the family Nectrinaceae (that comprise the genus Fusarium) was more abundant in asymptomatic than symptomatic cultivated plants and feral individuals (Figure S3), these differences however stayed non-significant according to Wilcoxon Rank Sum tests. Porras-Alfaro and Bayman (2007) previously reported Tulasnella, Ceratobasidium y Thanatephorus from microbiological isolates characterized by ITS Vanilla planifolia y poitaei. We recovered Ceratobasidium in our microbiological isolates in our live collection, and in the metabarcoding data.
{kind=link}