First, we need to choose an appropriate model chemistry which gives tautomer energies of a parent molecule as close as possible to experimental results. The method should not be very computationally expensive because our main subject – the Sildenafil molecule contains 63 atoms, thus the number of atomic orbital basis functions will be quite large, and the computations can be very time-expensive.
Choosing the model chemistry
We begin by evaluating the 2-hydroxypyridine/2-pyridone relative tautomer stability at various levels of theory and comparing them to the experimental results. First, the tautomers were optimized at the B3LYP/6-31G(d,p) level. We decided to use the 6-31G(d,p) basis set instead of standard 6-31G(d) because of the presence of additional polarization p-function for hydrogen atoms in the former, which is better suited for modelling the proton-transfer and prototropic tautomerism. The obtained enthalpy and Gibbs free energy are presented below.
Table 1. The enthalpy and Gibbs free energy of the 2-hydroxypyridine (2HP_C) and 2-pyridone tautomers (2HP_B) optimized at the B3LYP/6-31G(d,p) level of theory
|
Tautomer
|
ΔH
[kcal/mol]
|
ΔG
[kcal/mol]
|
ΔH
[kcal/mol]
|
ΔG
[kcal/mol]
|
|
|
Gas Phase
|
Water solution (PCM)
|
|
2HP_B
|
0.00
|
0.00
|
0.00
|
0.00
|
|
2HP_C
|
-0.11
|
0.03
|
3.56
|
3.60
|
|
exper.
|
0.46
|
|
3.4
|
|
It follows from Table 1 that this simple theoretical level agrees well with the experimental results, especially in the water solution. In the gas phase the order of tautomers is reversed but the enthalpy difference is very small. This can be of course because of a fortuitous cancellation of errors, so we followed with B3LYP calculations using more extended orbital basis sets. The results are gathered below in Table 2 where we give only the enthalpy as the focus is on comparison with experiment and the experimental data is enthalpy. We will follow the convention of subtracting the pyridone tautomer energy from hydroxypyridine tautomer energy in the following tables as the pyridone tautomers most often has lower energy, and operating the positive values is more convenient.
Table 2. The relative entalphy of the 2-hydroxypyridine/2-pyridone tautomers optimized at the B3LYP level of theory with various basis sets (ACCX = aug-cc-pVXZ)
|
|
6-31G(d,p)
|
6-311++G(d,p)
|
ACCD
|
ACCT
|
ACCQ
|
exper.
|
|
ΔH [kcal/mol]
Gas phase
|
-0.11
|
0.74
|
0.12
|
0.37
|
0.39
|
0.46
|
|
ΔH [kcal/mol]
Water solution (PCM)
|
3.56
|
5.27
|
4.71
|
4.91
|
4.91
|
3.40
|
It follows from Table 2 that larger basis sets give the proper tautomer order in the gas phase. In water solution modelled by PCM method the theoretical methods seems to overestimate the pyridone tautomer stability in comparison to experimental result. The results of calculations using the correlation-consistent basis sets behaves as expected with ACCT and ACCQ basis sets giving almost the same result. However even the simpler ACCD basis set gives a result within 0.2-0.3 kcal/mol from larger bases, thus for Sildenafil molecule he ACCD basis set should be of appropriate size. Therefore we decided that our method of choice for evaluation of Sildenafil molecule tautomer stability will be B3LYP/aug-cc-pVDZ.
We also tested the other DFT method, namely M06-2X, a compound method: CBS-QB3 and two ab initio methods: MP2, CCSD(T) (Single Point Energy). The results are compared below.
Table 3: The relative entalphy of the 2-hydroxypyridine/2-pyridone tautomers optimized at the various levels of theory and aug-cc-pVDZ (ACCD) basis set.
|
|
MP2
|
CBS-QB3
|
CCSD(T) SP
geom. B3LYP
|
CCSD(T) SP
geom. MP2
|
M06-2X
|
exper.
|
|
ΔH [kcal/mol]
Gas phase
|
-2.40
|
-1.41
|
-1.20
|
-1.37
|
-2.21
|
0.46
|
|
ΔH [kcal/mol]
Water environment (PCM)
|
1.75
|
2.77
|
2.92
|
2.69
|
2.21
|
3.40
|
Using the MP2, M06-2X, CBS-QB3 calculations the tautomers were fully optimized but in the case of CCSD(T) method the Single Point Energy calculation was performed on B3LYP and MP2 optimized geometries and the proper thermal corrections to enthalpy were added.
It follows that ab initio methods seem to overestimate the stability of enol (2-hydroxypyridine) tautomer in gas phase, a fact that is well known for the MP2 method and was mentioned in the Introduction. A similar problem is present with CBS-QB3 compound method and also with M06-2X DFT method. It is interesting to note that the M06-2X functional results are close to MP2 results. In the water solution the order of tautomers is at least proper, however we observe the same bias – the enol tautomer stability is overestimated by other methods comparing to B3LYP and experimental results.
The MP2, CCSD(T) and CBS-QB3 methods are too time expensive to employ them for such a large molecule as Sildenafil. As the M06-2X method gives similar results to them, but at a substantially lower computational cost, we decided to use it as a second method of choice and to compare its results to B3LYP results. Thus the second theoretical method used for the Sildenafil molecule will be M06‑2X/aug-cc-pVDZ.
Tautomerism of Sildenafil molecule
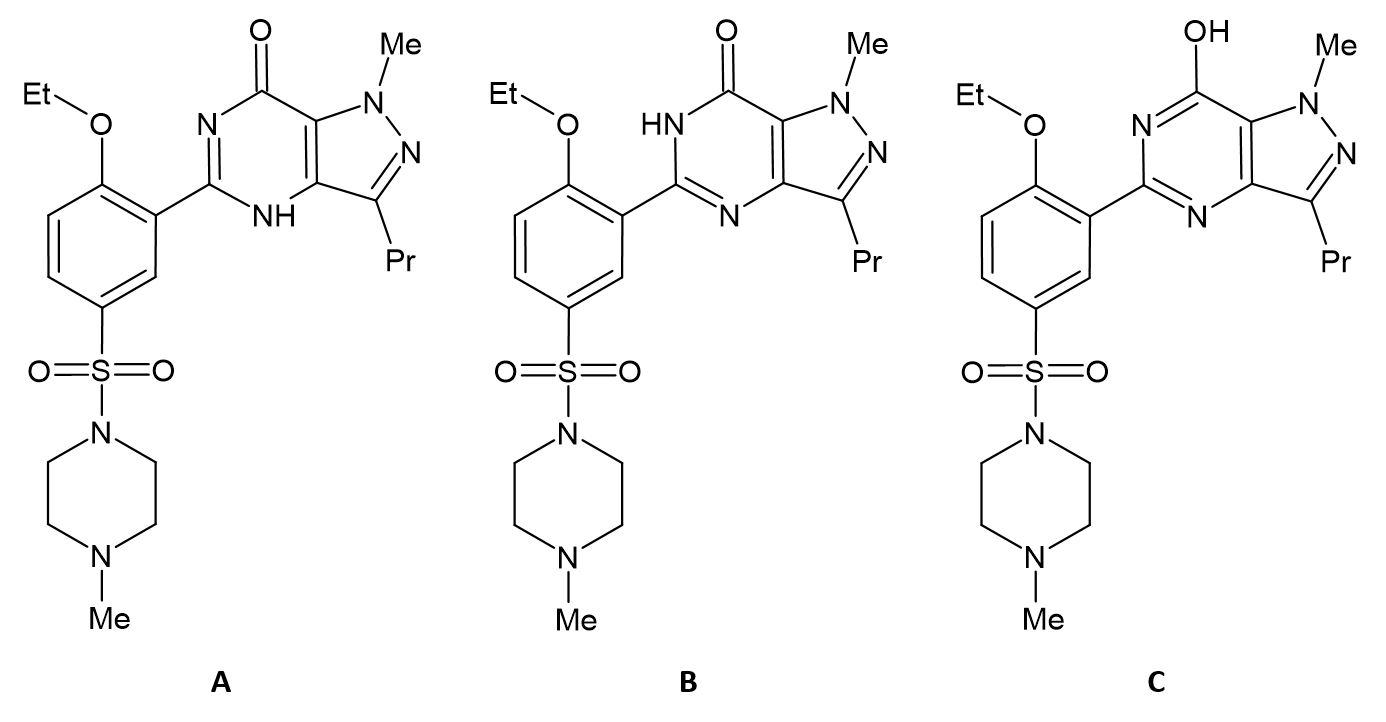
The initial structure of Sildenafil molecule was downloaded from the PubChem database[42]. This is the B type of pyrimidinone tautomer. The two other possible tautomeric forms A and C were built on the basis of the PubChem structure by proper shifts of the proton. Next, these three tautomers were optimized at the B3LYP/6-31G(d,p) level of theory in gas phase and water solution using the PCM model. The Gibbs free energy and relative energies of these structures are gathered below.
Table 4. The Gibbs free energy/relative energy of the Sildenafil tautomers optimized at the B3LYP/6-31G(d,p) level of theory in the gas phase and in water environment modelled by the PCM method
|
|
Gas phase
|
Water solution (PCM)
|
|
Tautomer
|
G [hartree]
|
ΔG [kcal/mol]
|
G [hartree]
|
ΔG [kcal/mol]
|
|
A
|
-1883.524084
|
11.48
|
-1883.553281
|
7.03
|
|
B
|
-1883.542373
|
0.00
|
-1883.564477
|
0.00
|
|
C
|
-1883.520329
|
13.83
|
-1883.542851
|
13.57
|
From these initial results we see that the keto B tautomer is most stable in both environments. The most striking feature is that the energy difference between it and the enol C tautomer is very large (13.83 kcal/mol) comparing to 4-hydroxypyrimidine/4-pyrimidinone (0.48 kcal/mol) mentioned in the Introduction. In water environment this energy gap stays at a very similar level. In the gas phase the energy of the A form is slightly lower than C so both A and C are not detectable in the tautomeric mixture. However, in water solution, the energy gap to A pyrimidinone form is lowered.
As the Sildenafil molecule contains some rotatable single bonds, it is necessary to perform a conformational analysis to obtain the lowest energy conformations of all tautomers. This should not have a large impact on the result, but is necessary to perform, if our aim is to obtain the real lowest energy tautomers. All three tautomers were thus subjected to systematic rotor search procedure, all single bonds were rotated and all possible conformers generated using the Molecular Mechanics MMFF force field. The resulting conformers were ordered by increasing energy and 5 lowest energy conformers for each tautomer were chosen for optimization at the B3LYP/6-31G(d,p) level in the gas phase. The Gibbs free energies of the optimized conformers are presented in Table 5 below.
Table 5. The Gibbs free relative energy of the lowest energy conformers of Sildenafil tautomers A, B, C reoptimized at the B3LYP/6-31G(d,p) level of theory in the gas phase. The “Starting structure” is the original structure before the conformational search. Lowest energy conformers are in bold.
|
Conformer
|
ΔG [kcal/mol]
|
|
A
|
B
|
C
|
|
1
|
0.58
|
0.66
|
0.75
|
|
2
|
0.02
|
0.61
|
0.00
|
|
3
|
0.34
|
0.51
|
0.31
|
|
4
|
0.00
|
0.00
|
0.81
|
|
5
|
0.32
|
0.10
|
0.06
|
|
Starting structure
|
0.59
|
0.11
|
0.30
|
Note that the order of conformers in the Table 5 is by the MMFF energy and it follows that according to B3LYP/6-31G(d,p) the order of stability is a little bit different. In each column in the last row the starting structure which was optimized before the conformational search is shown. It follows that for all three tautomers, thanks to conformational analysis, we obtained the lower energy conformers. The energy lowering is at a level of fraction of kcal/mol but nevertheless it exists. In the Table 6 below we gathered the lowest energy conformers of our 3 Sildenafil tautomers.
Table 6. The Gibbs free energy/relative energy of the Sildenafil tautomers optimized at the B3LYP/6-31G(d,p) level of theory in the gas phase
|
Tautomer
|
G [hartree]
|
ΔG [kcal/mol]
|
|
A_conf_4
|
-1883.525017
|
11.00
|
|
B_conf_4
|
-1883.542553
|
0.00
|
|
C_conf_2
|
-1883.520802
|
13.65
|
Comparing to Table 4 the absolute values of Gibbs free energy are lower, but relative values are similar because the energy lowerings were quite similar and thus they didn’t influence the relative energies much. Next, we reoptimized these best conformers at the B3LYP/aug-cc-pVDZ level of theory. The results are gathered in Table 7 below.
Table 7. The Gibbs free energy/relative energy of the Sildenafil tautomers optimized at the B3LYP/aug-cc-pVDZ level of theory in the gas phase and in water environment modelled by the PCM method.
|
|
Gas phase
|
Water solution (PCM)
|
|
|
Tautomer
|
G [hartree]
|
ΔG [kcal/mol]
|
G [hartree]
|
ΔG [kcal/mol]
|
|
A
|
-1883.676990
|
10.05
|
-1883.708749
|
5.49
|
|
B
|
-1883.693011
|
0.00
|
-1883.717503
|
0.00
|
|
C
|
-1883.674026
|
11.91
|
-1883.697599
|
12.49
|
|
|
|
|
|
|
It follows from Table 7 that at this level of theory the B tautomer is still dominant, however the energy gap to A and C is slightly lowered. For the water solution the similar trend is observed, the B tautomer is dominant but the gap to A is lowered by about 2 kcal/mol (compare Tables 7 and 4).
The 3d models of optimized Sildenafil tautomers are shown in Figure 1 below.
One striking structural feature is that in both A and B tautomers there exist an intramolecular hydrogen bond between the N-H of the pyrimidinone ring and O from the ethoxy group of the second phenyl ring. A second, much weaker hydrogen bond is formed by C-H of the phenole ring and the pyrimidine-type nitrogen atom of the pyrimidinone. The whole fragment of the Sildenafil molecule is appropriately rotated to facilitate the hydrogen bonds and thus the three rings lie approximately in one plane. The case of C tautomer is different. Only the weak C-H …. N hydrogen bond is possible but there is present an unfavourable steric interaction of oxygen and nitrogen atoms from both 6-membered rings which causes the rotation of the C-C bond connecting the rings and thus the two rings do not lie in one plane. It is a factor which certainly destabilizes the C tautomer comparing to A and B tautomers.
As a second method of choice we performed the M06-2X/aug-cc-pVDZ calculations for the lowest conformers of the Sildenafil molecule. The results are gathered in Table 8 below.
Table 8. The Gibbs free energy/relative energy of the Sildenafil tautomers optimized at the M06‑2X/aug-cc-pVDZ level of theory in the gas phase and in water environment modelled by the PCM method.
|
|
Gas phase
|
Water solution (PCM)
|
|
Tautomer
|
G [hartree]
|
ΔG [kcal/mol]
|
G [hartree]
|
ΔG [kcal/mol]
|
|
A
|
-1883.083989
|
9.12
|
-1883.116007
|
4.27
|
|
B
|
-1883.098517
|
0.00
|
-1883.122806
|
0.00
|
|
C
|
-1883.081624
|
10.60
|
-1883.106500
|
10.23
|
Comparing Tables 8 and 7 we see the trend previously observed for 2-hydroxypyridine tautomers that the M06-2X method slightly favours the enol tautomers both in gas phase and in water solution. Therefore, the energy difference between enol tautomer and keto tautomers is lowered by about 2 kcal/mol. Also the energy difference between two keto tautomers is lowered by about 1 kcal/mol.
Tautomerism of the model in-between molecules
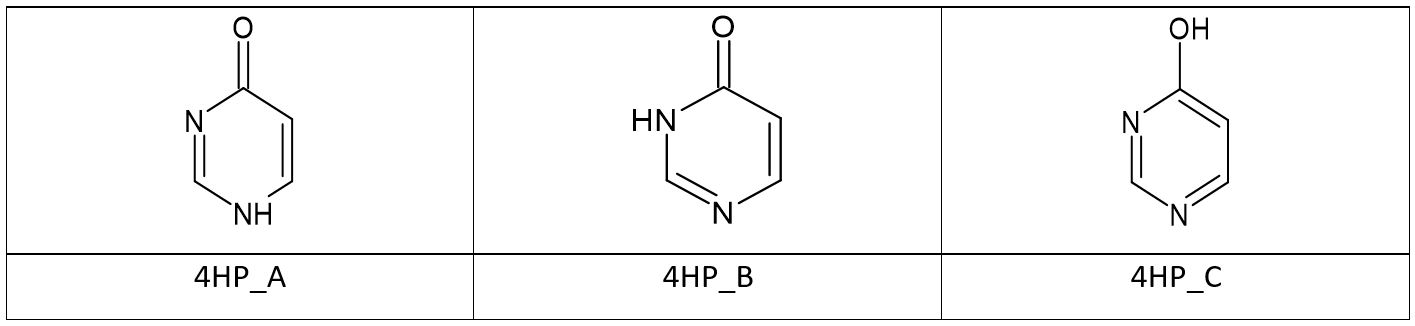
To look more deeply into the reasons behind the relative energy differences of A, B and C tautomers we decided to build and optimize model molecules which are in-between 4-hydroxypyrimidine/4-pyrimidinone and Sildenafil and to compare the relative tautomer energies at each step. As we need only the qualitative explanation the simple B3LYP/6-31G(d,p) model was chosen for these calculations. We decided to use the following model molecules:
Thus first we observe how the fusion of the pyrazole ring influences the tautomeric equilibrium of 4-hydroxypyrimidine, second we observe how the addition of the ethoxy-phenyl ring affects the equilibrium and at the end we observe the effect of addition of the rest of the Sildenafil molecule. The results obtained are gathered in Table 9.
Table 9. The Gibbs free energy [kcal/mol] of the model in-between and Sildenafil tautomers optimized at the B3LYP/6-31G(d,p) level of theory in the gas phase
|
Tautomer
|
4HP
|
4HP-pz
|
4HP-pz-PheEt
|
Sildenafil
|
|
A
|
9.62
|
10.32
|
9.40
|
11.00
|
|
B
|
0.00
|
0.00
|
0.00
|
0.00
|
|
C
|
1.29
|
7.55
|
13.60
|
13.65
|
The results are shown also in Figure 3 to better illustrate the trends in the energy change.
The difference of energy between A and B tautomers is changing not very much. It basically oscillates within 10 kcal/mol for all studied molecules. A very different picture emerges for C and B relative energy. For the starting 4-hydroxypyrimidine/4-pyrimidinone system this energy gap is very small. Experimentally it is determined as 0.48 kcal/mol and theoretically estimated by us at 1.29 kcal/mol. Thus these two forms coexist in equilibrium. However even first step – fusion with pyrazole ring dramatically influences this equilibrium by strongly disfavouring the C form. For the 4HP-pz system the energy gap is 7.55 kcal/mol and the C tautomer is already almost not present in the tautomeric mixture. The reason for this behaviour can be associated with changes of aromaticity. The 6-membered ring of the C form is highly aromatic comparing to the B form. A very rough and approximate measure of this aromaticity can be drawn from the picture of the “aromatic” bonds which order is between of 1 and 2. This is nicely illustrated in Figure 2: almost all the ring bonds of 4HP_C are “aromatic” but only two of them in 4HP_B. Now, let us compare the 4HP-pz molecules (after the fusion with pyrazole): the 4HP-pz_C tautomer lost one “aromatic” bond and the 4HP-pz_B tautomer got two additional “aromatic” bonds. Thus we may expect increase of aromaticity of the B tautomer (stabilization) and decrease of C (destabilization).
The next step – addition of the phenyl ring with the etoxy group enlarges the energy gap between C and B tautomers up to 13.6 kcal/mol. This time the reason is a strong intramolecular hydrogen bond present in B (and also in A) but not present in C. Additionally in the C tautomer there exist an unfavourable steric repulsion between O (ethoxy group) and N (6-membered ring) atoms.
In order to deeper understand the trends presented in Table 9 and Figure 3 we performed the aromaticity estimation using two indices: geometric HOMA and electronic pEDA. The results are presented in Tables 10 and 11.
Table 10. HOMA index of various parent molecules of Sildenafil.
|
Nazwa
|
4HP
|
4HP-pz
|
4HP-pz-PheEt
|
Sildenafil
|
|
5-ring
|
6-ring
|
5-ring
|
6-ring
|
5-ring
|
6-ring
|
5-ring
|
6-ring
|
|
A
|
-
|
0.415752
|
0.915586
|
0.587573
|
0.907504
|
0.649285
|
0.906729
|
0.644629
|
|
B
|
-
|
0.627920
|
0.875378
|
0.678504
|
0.862715
|
0.730823
|
0.859539
|
0.727147
|
|
C
|
-
|
0.988619
|
0.799357
|
0.937675
|
0.794111
|
0.939296
|
0.792986
|
0.938766
|
The value for 5-membered ring for 4HP is absent because there is no 5-membered ring in this molecule. In order to get reference the indices of a free 5-membered ring we optimized the N-methylated 3-Me-pyrazole – see Figure 3. The aromaticity indices for this molecule are as follows: HOMA = 0.8906 and pEDA = -0.01782
The geometric HOMA index is based on the length of the bonds. Basically HOMA index tends to unity when the bond lengths of the ring are all close to the “aromatic” bond length similar to that in benzene. On the contrary, if the bonds are pure single and double, the HOMA tends to zero. As we see in Table 10 for 4HP molecule, the HOMA for C tautomer is quite close to 1, thus is highly aromatic. The B tautomer is much less aromatic and the A tautomer is even less aromatic. If we take a look at the Figure 2 we see that in case of B tautomer the two “aromatic” bonds are separated by on double bond, and in the A tautomer the both are connected to the same pyrrole-type nitrogen atom. Apparently the latter is regarded by HOMA algorithm as less aromatic. Fusion with pyrazole ring is reflected by de-aromatization of the C ring (from 0.99 to 0.94) and aromatization of the A (from 0.42 to 0.59) and B (0.63 to 0.68) rings. The aromaticity of A and B rings are now much more similar, however still the B tautomer is more aromatic. Addition of Ethoxy-phenyl ring still increases aromaticity of A and B tautomers, but do not lower that of C tautomer and the final addition of the rest of the Sildenafil molecule do not alter the situation much.
As far as the 5-membered (pyrazole) ring is concerned, we see that for the 4HP-pz molecule it loses its aromaticity for B tautomer and especially for C, but slightly increases for A. The aromaticity of the 5-membered ring do not change much for the consecutive, more complex molecules.
Table 11. pEDA index of various parent molecules of Sildenafil
|
Nazwa
|
4HP
|
4HP-pz
|
4HP-pz-PheEt
|
Sildenafil
|
|
5-ring
|
6-ring
|
5-ring
|
6-ring
|
5-ring
|
6-ring
|
5-ring
|
6-ring
|
|
A
|
-
|
0.56567
|
-0.06294
|
0.74448
|
-0.06352
|
0.76148
|
-0.07174
|
0.76116
|
|
B
|
-
|
0.50103
|
-0.12566
|
0.7310
|
-0.11995
|
0.74241
|
-0.13427
|
0.74424
|
|
C
|
-
|
0.14731
|
-0.14848
|
0.35856
|
-0.13991
|
0.37776
|
-0.15528
|
0.37860
|
Regarding the pEDA index it is defined here as the electron excess/deficiency from the ideal Huckel sextet (6 pi electrons). Thus we see that for the 4HP molecule we observe a slight excess for highly aromatic C tautomer (only 0.15e) and much larger excess for A and B molecules (more that 0.5e). Such large pi-electron excess means lower aromaticity and this picture is in accordance to HOMA – the B tautomer is more aromatic that A. After fusion with pyrazole the pi-electron excess of C tautomer is increased and this is in accordance with decrease of HOMA. However for A and B tautomers pEDA indicates similar increase of pi-electron excess, therefore we see that the fusion with pyrazole increase the pi-electron excess for all tautomers. As it follows from Table 11 for the rest of the molecules there is no great change in pi-electron excess of the 6-membered ring.
Regarding the 5-membered ring it becomes more pi-deficient which can be especially seen for B and C (the largest change) tautomers.
{kind=link}
{kind=link}
{kind=link}