EVA swab material selection
Validation of the macrofoam swabs (EVA swab tool material) to collect microorganisms from various material types was not a part of this study. However, a comprehensive study was performed previously to understand the suitable swab materials (cotton, polyester, and macrofoam) in the efficient removal of the microorganisms from the aluminum and titanium surfaces [21]. Briefly, a model microbial community comprised 11 distinct species of bacterial, archaeal, and fungal lineages, was used to examine the effects of variables in sampling matrices, target cell density/molecule concentration, and cryogenic storage on the overall efficacy of the sampling regimen. The biomolecules and cells/spores recovered from each collection device were assessed by cultivable and microscopic enumeration, and quantitative and species-specific PCR assays. rRNA gene-based quantitative PCR analysis showed that cotton swabs were superior to nylon-flocked swabs and macrofoam swabs significantly outperformed polyester wipes. Furthermore, macrofoam swab materials were found to withstand extreme temperature fluctuations of the space conditions including varying pressure, and vacuum [5].
EVA swab sample kit preparation and sample collection
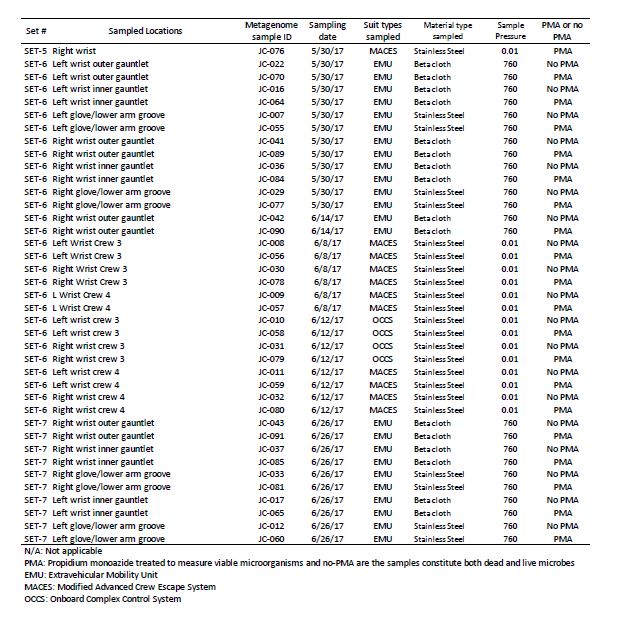
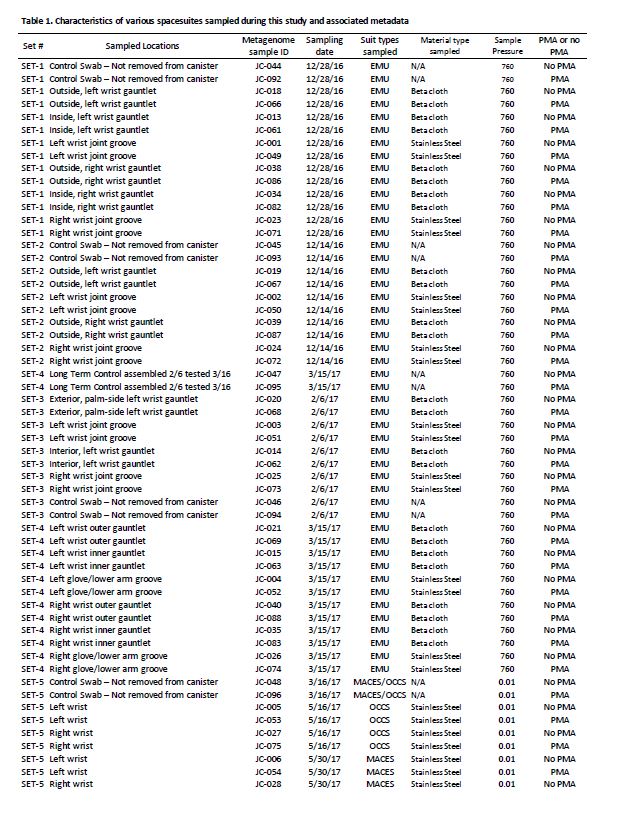
Three different kinds of spacesuits were sampled (Figure 1). Briefly, the EMU suits are currently used for EVA on ISS, but are not planned for use in future missions. We sampled stainless steel wrist joints and cloth gauntlets covering the joints on these suits. The outer fabric of the EMU is made of Ortho-Fabric, which is a blend of Gortex (ePTFE), Kevlar (a para-aramid type fiber related to nylon) and Nomex (a meta-aramid type fiber)[22]. The MACES and OCSS suits designed for high-risk parts of a mission, such as inside Orion during launch and reentry through Earth’s atmosphere, use the same wrist joint as the EMU but without a gauntlet to cover it. The outer layer of the MACES and OCSS suits is comprised of orange Nomex [23, 24]. NASA has conducted a series of ground tests intended to evaluate the EVA swab kit’s form, fit, and function under mission operations scenarios, in preparation for eventual sample collection from outside the ISS [5]. For samples collected from the EMU, EVA swabbing was an add-on to a routine suit familiarization test that all flight crew are required to perform. Familiarization involves suit fit and functional checks, followed by a four-hour prebreathe protocol (to mitigate potential for decompression sickness) before exposure to vacuum in the Space Station Airlock Test Article chamber. Spacesuit samples were collected during the prebreathe protocol, when the crew member was breathing pure oxygen at a suit internal pressure 4.3 psi higher than ambient external pressure, but not yet at external vacuum pressure. Although standard laboratory swabs could have been used under these conditions, this test provided an opportunity for suited crew to practice self-swabbing with the flight-like EVA swab kit, which will be necessary in future studies where samples will be taken under external vacuum conditions. A second series of tests was conducted with the MACES and OCSS suits. In these tests, four test subjects sampled their own suits (two MACES suits and two OCSS suits) inside the 11-foot vacuum chamber while the chamber was at vacuum (0.01 torr). The internal pressure inside the suits was 4.3 psi. Samples collected during these tests were exposed to four hours of vacuum.
Sample kit cleaning, sterilization, and assembly were performed at JSC according to a purpose-developed protocol. Each sample canister (assembled with filter and ball plungers) and swab end effector assembly was placed into separate autoclave bags. Bagged components were placed into Steris LV 250 laboratory steam sterilizer and sterilized using a gravity cycle of 45 minutes at 121˚C (250˚F) and 103.4 kPa (15 psi). Note that neither the sample caddy itself nor the tool handle was autoclaved. Bagged components were allowed up to 1 hour of cool-down time at approximately 22˚C (71.6˚F) for safe handling. Following autoclaving, bagged components were transferred to a Labconco Horizontal Clean Bench (Model # 36100000, ISO Class 5). With the commercial swab inside its sterile packaging, the swab stem was cut to optimal length (approximately 6.0 cm (2.4 in)) using sterilized wire cutters, making sure the swab head remained inside its packaging until the final assembly step. The cut end of the swab was then inserted into the end effector slot, and set screws were tightened to hold the swab in place. Sterile packaging was removed from the swab head immediately before inserting each swab assembly into its sterile container. Each container/swab assembly was then mounted into the tool caddy, wiped clean with isopropanol and placed into bonded storage until the test.
During swab assembly, technicians wore sterile gloves, and both the gloves and assembly tools (Allen wrench, scissors, and forceps) were sprayed with 70% ethanol surface disinfectant. All parts were handled either with sterile forceps or the autoclave bags, with no contact between the gloves and tool areas that must remain sterile. After assembly, the EVA sample kits were transported to the test site packed inside hard-sided storage cases. Once at the test site, the analog crew were briefed on tool usage and were given an opportunity to practice with a spare handle and sample caddy assembly. Over a period of seven months between Dec 2016 and June 2017, 176 spacesuits, environmental control, and floor samples were collected during 8 sampling time periods at JSC. Figure 1 shows sample collection from various parts of the spacesuits, EVA sampling kits, and number of samples associated with various spacesuits. In this study, among 176 samples, 48 spacesuits including five controls samples were further analyzed for various microbiological characteristics using traditional and shotgun metagenomic sequence analyses. Environmental controls were the swabs that were removed from the canister during testing but not touched to any surface. Negative controls were swabs that were not opened at all during testing. Among these 48 samples, 36 were from EMU and 12 were from MACES spacesuits. The specific location for each sampling event of these 48 samples, surface area, and collection dates are given in Table 1 and detailed metadata about spacesuit types used, fabrics composition, microbial burden, cultivable diversity, are given in the Supplementary Table S1.
Sample processing
After sample collection, sample processing took place in an ISO 7 (10K class) cleanroom at JPL. Under ISO 5 certified biosafety cabinet, each swab was aseptically severed with a sterile cutter and transferred to a 50 mL Falcon tube containing 15 mL of sterile phosphate-buffered saline (PBS; pH 7.4). The tube with the swab was shaken for two minutes followed by concentration with a Concentrating Pipette (Innova Prep, Drexel, MO) using 0.22 µm Hollow Fiber Polysulfone tips (Cat #: CC08022) and PBS as elution fluid. Each sample was concentrated to 5 mL. A 100 µL concentrated aliquots were plated on various agar plates to estimate cultivable population using traditional plate count methods (described below). One mL of the diluted solution (200 µL plus 1.8 mL PBS) was used to conduct an ATP assay (Kikkoman Corp., Noda, Japan) to rapidly measure total and viable microbial population [25], enabling appropriate serial dilutions. Furthermore, 3 mL of each concentrated sample was split into two 1.5 mL- aliquots and one aliquot was treated with PMA to assess viability [19], while the second aliquot was handled similarly but without the addition of PMA. Briefly, 18.25 µL of 2 mM PMA was added to one half of the 3-mL sample (final concentration 25 µM) followed by 5 min incubation at room temperature in the dark and 15 min exposure to the activation system (PMA LED device, Biotium, Hayward, CA). Each sample was then split into two 0.75 mL aliquots. One aliquot was transferred to bead beating tubes containing Lysing Matrix E (MP Biomedicals, Santa Ana, CA), followed by bead beating for 60 s using the vortex sample holder (MO Bio, Carlsbad, CA). The bead-beaten aliquot and the aliquot without bead beating were combined for their corresponding PMA-treated and non-treated samples. DNA extraction was accomplished with the Maxwell 16 automated system (Promega, Madison, WI), in accordance with manufacturer instructions. A Maxwell control (MC) without any sample added in its cartridge was run concurrently with each flight sample set to account for microbial contamination associated with reagents (kitome) used in the automated DNA extraction. The extracted DNA was eluted in 50 μL of water and stored at −20˚C and processed with the rest of the samples later.
Estimation and identification of cultivable microbial population
The 100 µl of each concentrated sample were plated on Reasoner's 2A agar (R2A for environmental microbes), Potato Dextrose Agar with chloramphenicol (100 µg/mL; PDA for fungi), and blood agar (BA for human commensals; Hardy Diagnostics, Santa Maria, CA) in duplicate. R2A and PDA plates were incubated at 25˚C for seven days and BA plates at 37˚C for two days at which time colony forming units (CFU) were counted. All colonies were picked from each plate and from each suit sampling location. The isolates were then archived in semisolid R2A or PDA slants (agar media diluted 1:10) and stored at room temperature. Once a culture was confirmed to be pure, two cryobead stocks (Copan Diagnostics, Murrieta, CA) were prepared for each isolate and stored at –80˚C. A loopful of purified microbial culture was directly subjected to PCR, and the targeted fragment was amplified (colony PCR), or DNA was extracted with the UltraClean DNA kit (MO Bio, Carlsbad, CA) or Maxwell 16 instrument. The extracted DNA was used for PCR to amplify the 1.5 kb 16S rRNA gene to identify bacterial strains. The following primers were used for the 16S rRNA gene amplification to estimate bacterial population. The forward primer, 27F (5’-AGA GTT TGA TCC TGG CTC AG-3’) and the reverse primer, 1492R (5’-GGT TAC CTT GTT ACG ACT T-3’) [26, 27]. The PCR conditions were as follows: denaturation at 95 ˚C for 5 min, followed by 35 cycles consisting of denaturation at 95 ˚C for 50 s, annealing at 55 ˚C for 50 s, and extension at 72 ˚C for 1 min 30 s and finalized by extension at 72 ˚C for 10 min. For fungal population estimation, the forward primer ITS 1F (5’- TTG GTC ATT TAG AGG AAG TAA-3’) [28] and reverse primer Tw13 (5’-GGT CCG TGT TTC AAG ACG-3’) [29] were used to obtain ~1.2 kb ITS product. The PCR conditions were as follows: Initial denaturation at 95˚C for 3 min followed by 25 cycles of 95 ˚C for 50 s, annealing at 58 ˚C for 30 s, and extension at 72 ˚C for 2 min, followed by a final extension at 72 ˚C for 10 min. The amplicons were inspected by gel electrophoresis in 1% agarose gel. When bands for products were visible, amplification products were treated with Antarctic phosphatase and exonuclease to remove 5’- and 3’- phosphates from unused dNTPs before sequencing. The sequencing was performed (Rockville, MD, USA) using 27F and 1492R primers for Bacteria, and ITS1F and Tw13 primers for fungi. The sequences were assembled using SeqMan Pro from DNAStar Lasergene Package (DNASTAR Inc., Madison, WI). The bacterial sequences were searched against EzTaxon-e database [30] and the fungal sequences against the UNITE database [31]. The identification was based on the closest percentage similarity (>97%) to previously identified microbial type strains.
qPCR assay
Following the DNA extraction, quantitative polymerase chain reaction (qPCR), targeting the partial 16S rRNA gene (bacteria) or partial ITS region (fungi), was performed with SmartCycler (Cepheid, CA) to quantify the microbial burden as previously established [32]. Each 25-μL reaction consisted of 12.5 μL of 2X iQ SYBR Green Supermix (BioRad, Hercules, CA), 1 μL each of forward and reverse oligonucleotide primers (10 μM each), and 1 μL of template DNA (PMA treated and non-treated samples). Each sample was run in triplicate; the average and standard deviation were calculated based on these results. Purified DNA from a model microbial community [21] served as the positive control and DNase/RNase free molecular-grade distilled water (Promega, Madison, WI) was used as the negative control in each run. The number of gene copies was determined from the standard curve as described previously with a modification where synthetic fragments of B. pumilus (1.4 kb 16S rRNA gene) or Aureobasidium pullulans (1-kb ITS region) were used instead of genomic DNA [33]. The qPCR efficiency was ~98% The negative control values were not deducted since the values were at ~100 copies per 1 or 10 µL and not scalable (yielded the same results despite using 1 µL and 10 µL of DNA templates).
Illumina based DNA sequencing and analysis
The initial DNA yield and metagenome library quantitation of all 96 samples tested (48 samples PMA treated and 48 samples PMA untreated) were measured by Qbit (Thermo Fisher Scientific Inc., USA). DNA libraries for all 96 samples were prepared for shotgun metagenome sequencing using the Nextera DNA Library Preparation Kit from Illumina. The quality and fragment size of each library were assessed on a Bioanalyzer 2100 (Agilent). Separate adapters were added to the DNA from each library, normalized to 2 nM, pooled, denatured, and diluted to 1.8 pM according to the standard recommendations by Illumina. The HiSeq4000 platform (Illumina) was used for sequencing, resulting in 100-bp paired-end reads.
Bioinformatics Analysis
Bioinformatic analyses were performed on Weill Cornell Medicine's Athena compute cluster, a typical high-performance grid compute (Slurm) system. The secondary analysis was performed on Linux and MacOS systems. Unless otherwise noted programs were run with default settings.
Data Quality Control and Filtering
Sequence data were processed with AdapterRemoval (v2.17) to remove low-quality reads and reads with ambiguous bases. Subsequently, reads were aligned to the human genome (hg38, including alternate contigs) using Bowtie2 (v2.3.0, fast preset). Read pairs where both ends mapped to the human genome were separated from read pairs where neither mate mapped. Read pairs where only one mate mapped were discarded. Hereafter, we refer to these read sets as human reads and non-human reads. We did not process human reads beyond counting the total fraction of DNA from our samples which mapped to the human genome.
Taxonomic Profiling and Analysis
Taxonomic profiles were generated by processing non-human reads KrakenUniq (v0.3.2) with a database based on all draft and reference genomes in RefSeq Microbial (bacteria, fungi, virus, and archaea) ca. March 2017. KrakenUniq uses a k-mer based approach to identify reads. Reads are broken into k-mers of 31 bases. Each k-mer is mapped to a database that lists the lowest common ancestor of all genomes which contained the k-mer. Each read is classified by identifying the best supported path in the taxonomic tree of markers. Finally, the taxonomic makeup of a sample is given by concatenating annotations for reads without further processing. KrakenUniq counts the number of unique marker k-mers assigned to each taxa, and we filtered taxa with fewer than 512 unique markers. Differential abundance estimation (where applicable) using the ALDEx2 R package was performed [34]. Briefly, ALDEx2 transforms read count matrices using a centered log ratio transformation that models samples as Dirichlet-Multinomial distributions over taxa then compares taxonomic abundances across groups. If two groups are given, comparison is done with a Wilcoxon rank sum test, more than two groups are tested via a generalized linear model. All p-values are multiple hypotheses corrected using Benjamini-Hochberg. We considered a taxon to have differential abundance in a given condition if its corrected p-value was less than or equal to p = 0.05.
Dimensionality reduction of taxonomic profiles was performed with Uniform Manifold Approximation and Projection UMAP [35] based on a matrix of Jensen-Shannon Divergences (JSD) between samples. Analysis of intersample diversity (beta-diversity) was achieved using the same matrix of JSD. Intrasample diversity (alpha-diversity) was measured by taking Shannon’s Entropy of the total sum normalized taxonomic profile of each sample. Rarefaction analysis of taxa was performed by grouping samples by location and setting and selecting 16 uniform random groups for each value. A curve of best fit was found by fitting a logarithmic model to the series.
Profile of Eukaryotic species were generated using CLARK-S (v1.2.5)[36] using sequences classified with high confidence (i.e., confidence score > 0.75, and gamma score > 0.10) as defined in the CLARK manual. Identification of taxa was further restricted to species with relative abundance at least 0.01 % of the total sequences.
Samples were compared to 8 representative samples of human body sites selected from the Human Microbiome Project (HMP)[37] for each of 5 body sites: oral, skin, airways, gastrointestinal, and urogenital. Using MetaPhlAn2 (v2.2)[38], we generated taxonomic profiles for HMP samples and our samples and compared profiles using Cosine Similarity.
Functional Profiling and Analysis
HUMANn2 [39] was used to generate functional metabolic profiles of the genes in our samples. Non-human reads were aligned to Uniref90 (ca. March 2017) using the DIAMOND aligner (v0.8.6)[40]. Subsequently, alignments were processed using HUMANn2 (v0.11.1) to produce profiles of pathway abundance. Pathways were tested for differential expression using the Wilcoxon rank sum corrected by Benjamini Hochberg. Dimensionality reduction of pathways was performed using PCoA over normalized pathway abundances.
Profiling Antimicrobial Resistance Genes
Profiles of antimicrobial resistance (AMR) genes using MegaRes (v1.0.1)[41] were created. To generate profiles from MegaRes, we mapped non-human reads to the database using Bowtie2 (v2.3.0, very-sensitive presets)[42]. Subsequently, alignments were analyzed using ResistomeAnalyzer (commit 15a52dd)[43] and normalized by total reads per sample and gene length to give Reads per kilo base per million mapped reads (RPKMs). MegaRes includes an ontology grouping resistance genes into, gene classes, AMR mechanisms, and gene groups.
Identification of Genomes and Strains
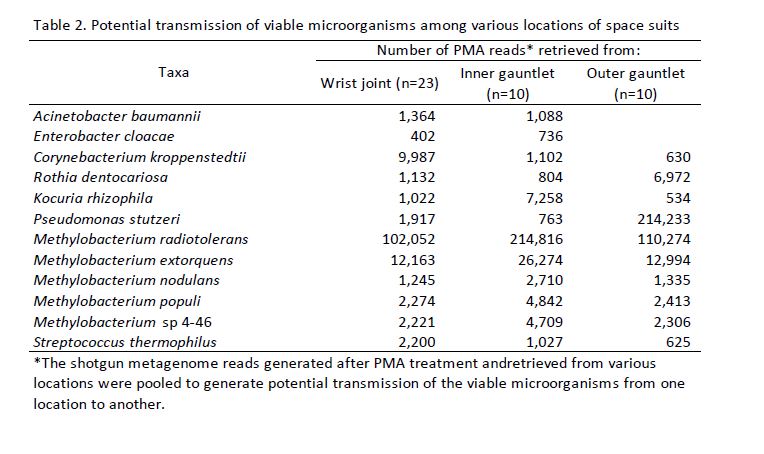
We assembled contigs from all PMA treated samples using MegaHIT (v1.1.3)[44] then clustered the resulting contigs into draft genomes using MetaBAT2 [45]. Draft genomes were quality controlled and assigned a rough taxonomic rank using CheckM [46]. Genomes with less than 50% completeness or more than 20% contamination were discarded. We aligned all genomes to one another to using Nucleotide MUMmer [47] and processed the results to generate an Average Nucleotide Identity (ANI) between all pairs of draft genomes. We discarded all alignments that covered less than half the average lengths of the genomes being aligned. We further discarded alignments with less than 99% ANI so that we would only be left with pairs of nearly identical genomes. We grouped these alignments into connected components and analyzed the sites where each component was found.
{kind=link}
{kind=link}
{kind=link}