Reagents and chemicals
Manganese chloride (MnCl2·4H2O) and actinomycin D, and other chemical compounds were bought from Sigma Chemical Co. (Saint Louis, MO, USA). Cell Counting Kit-8 (CCK-8) was supplied with Transgen Institute of Biotechnology (Beijing, China). FD Rapid GolgiStain™ kit was supplied by FD NeuroTechnologies (Ellicott City, MD, USA). Enzyme-linked immunosorbent assay (ELISA) kits of mice DA was acquired from Baolai Biotechnology Institute (Jiangsu, China). Ethyl ester derivative of MA (MA2, the FTO inhibitor) was a present from Professor Caiguang Yang of the Chinese Academy of Sciences. Fluoro-Gold (FG) was purchased from Kaiji Biotechnology Co., Ltd. (Jiangsu, China). The adeno-associated virus 5 (AAV5)-FTO and AAV5-ephrin-B2 were purchased from Genecreate Biological Co., Ltd. (Wuhan, China). Lentiviral particles for over-expression of FTO (FTO-OE) were purchased from Hesheng Gene Technology Co., Ltd. (Beijing, China). Anti-m6A antibody was bought from Synaptic Systems (USA, 202003). Protein-A bead was bought from Thermo Fisher (USA, 10002D). RNasin was provided by Promega Biotechnology Co., Ltd. (Beijing, China). Rabbit antibody of FTO, methyltransferase like 3 (METTL3), α-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5), YTH domain-containing family proteins 1/2 (YTHDF1/2) were bought from Proteintech Group, Inc (Wuhan, China). Rabbit antibody of tyrosine hydroxylase (TH) was bought from Cell Signaling Technology, Inc. Rabbit antibody of METTL14, ephrin-A1/3, ephrin-B1/3 were sold from ABclonal Biotechnology Co., Ltd. (Wuhan, China). Rat antibody of dopamine transporter (DAT), mouse antibody of ephrin-A5, ephrin-B2, Dopamine- and cAMP-regulated neuronal phosphoprotein 32 kDa (DARPP32) and GADPH were offered from Santa Cruz Biotechnology, Inc (Santa Cruz, CA, USA). Mouse antibody of ephrin-A2/4 was bought from Boaosen Biotechnology Co., Ltd. (Beijing, China). Horseradish peroxidase (HRP) conjugated anti-rabbit, mouse and rat secondary antibody were provided by means of Boaosen Biotechnology Co., Ltd. (Beijing, China). Real-time quantitative PCR (RT-qPCR) kit and Trizol reagent were supplied by TaKaRa Biotechnology Co, Ltd (Dalian, China). Other chemical substances were supplied through neighborhood chemical suppliers. All chemical substances reagents have been analytical grade or the pharmaceutical grade.
Animals
The research was carried out on adult C57BL/6 mice (20 ± 2 g; N = 350; Among them, 30 pregnant mice are needed for primary neuron culture) which have been bought from the Laboratory Animal Centre of China Medical University, Shenyang, China (SPF grade, certificate no. SCXK2013-0001). The mice were housed in plastic cages with 12-h light/dark cycle (temperature, 25 oC±1 oC, humidity, 55%±5%). Water and Food were accessible to the mice. All experiments were performed following the care and use guidelines of laboratory animals by Institutional Animal Care and Use Committee of China Medical University. The study protocol conformed to the National Institutes of Health Guide for Care and Use of Laboratory Animals (NIH Publications No. 8023, revised 1978). Attempts were made to minimize the number of animals used and to reduce animal suffering.
Primary neuron and SH-SY5Y culture
Neonatal C57 BL/6 mice were decapitated with scientific scissors without use of an anesthetic. The cranium was opened with ophthalmic forceps after sterilization with 75% alcohol. D-Hank's buffer solution was added to the dissected striatal tissue, and routinely separated with surgical devices under a light microscope. The tissue was digested with 0.125% trypsin solution for 20–30 min, and diluted with Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich, USA) containing 20% fetal bovine serum (FBS). Isolated neurons were seeded into 60-cm2 plastic tissue culture flasks precoated with poly-L-lysine after purification by filtration through a 200-mesh sieve. Cultures were maintained in 5% CO2/95% air at 37°C. After cell attachment, DMEM medium was replaced with Neurobasal™-A Medium supplemented with B27 (Life Technologies). Neurons were further purified by addition of cytosine arabinoside and cultured for 24 h (5 µM, Sigma-Aldrich). Primary cultures yielded more than 95% neurons, which was determined through NeuN antibody immunostaining after culturing for approximately 7 days.
SH-SY5Y cells were purchased from the National Centre for Cell Sciences of China. Cells were maintained in 5% CO2/95% air at 37°C and cultured with DMEM supplemented with 10% FBS, streptomycin, and penicillin.
Animal experiments
One hundred forty mice were randomly assigned to sixteen groups based on their weight, with 12 mice in each group. The first part included the control group, 12.5, 25, and 50 mg/kg MnCl2 groups. The second part included the AAV5 negative control (NC) group, AAV5-FTO control group, AAV5-NC + 50 mg/kg MnCl2 group, and AAV5-FTO + 50 mg/kg MnCl2 group. The third part included the control group, 20 mg/kg MA2 control group, 50 mg/kg MnCl2 group, and 20 mg/kg MA2 + 50 mg/kg MnCl2 group. The fourth part included the AAV5-NC group, AAV5-NC + 50 mg/kg MnCl2 group, AAV5-NC + 20 mg/kg MA2 + 50 mg/kg MnCl2 group, and AAV5-ephrin-B2 + 20 mg/kg MA2 + 50 mg/kg MnCl2 group. Mice in the control group received a subcutaneous (s.c.) injection of 0.9% NaCl and intraperitoneal (i.p.) injection of 0.9% NaCl after 2 h. Mice in the MnCl2 groups received s.c. injections of 0.9% NaCl and i.p. injections of 12.5, 25, and 50 mg/kg MnCl2 after 2 h. Mice in the MA2 control group were administered with s.c. injections with 20 mg/kg MA2, and i.p. injections of 0.9% NaCl after 2 h. Mice in the 20 mg/kg MA2 + 50 mg/kg MnCl2 group received s.c. injections with 20 mg/kg MA2, and i.p. injections of 50 mg/kg MnCl2 after 2 h. The concentration of treatments were 5 mL/kg. Treatments were administered daily two weeks.
For AAV5 viral injection, mice were anaesthetized with 5% chloral hydrate and positioned on a stereotaxic apparatus. Bilateral holes were drilled into the cranium at 0.5 mm foreside and ˗2.8 mm outside the bregma for administration of treatments into the striatum regions. An AAV5 expressing either FTO or ephrin-B2 (AAV5-FTO or AAV5-ephrin-B2) or hrGFP (AAV5-NC) was obtained through stereotactic injection. A glass cannula stuffed with a virus was placed on the striatum (-3.5 mm), and 0.2 µL virus was administered at a rate of 0.1 µL/min. The scalp was then sealed. Follow-up exposure was carried out 10 days after virus injection, and then behavioral experiments or other tests were carried out.
After the behavioral experiment, all the mice were sacrificed through decapitation under anesthesia. Striatum of 20 mice in each group was separated in an ice bath. 4 out of the 20 mice were used for histological and ultrastructural analysis of the striatum. Among the other 16 mice from each group, 4 were used for the detection of Mn and DA levels. 2 were used for immunofluorescence detection. 2 were used for determination of ephrin-B2 m6A mRNA expression, 2 for electrophysiological recording, and 2 mice were used for fluorogold retrograde tracing. Striatum for the other mice was used for determination of target mRNA level, protein, and other indicator levels.
Lentiviral generation and infection.
Lentiviral particles for over-expression of NC and FTO were purchased from Hesheng Gene Technology Co., Ltd. (Beijing, China). For over-expression of ephrin-B2, METTL3, METTL14, YTHDF1 or YTHDF2, cells were transfected with plasmids using lipofectamine 2000 (Sigma, USA) following the manufacturer’s instructions. After 48–72 h of transfection, the virus-containing supernatant was obtained and used to infect target cells, followed by screening with puromycin (Solarbio, 2.5 µg/mL) for 7 days.
Cells treatments
Cells were cultured in media for 24 h with 0, 125, 250, and 500 µM MnCl2. In the 0 and 500 µM MnCl2 groups, cells were used for over-expression of FTO, ephrin-B2, YTHDF2, FTO + METTL3 + METTL14, FTO + ephrin-B2, and FTO + YTHDF1 or YTHDF2. In addition, For the MA2 intervention groups, cells were cultured in media for 24 h with 0 µM MnCl2, 200 µM MA2, 500 µM MnCl2, and 200 µM MA2 (pretreated for 6 h) + 500 µM MnCl2. The relevant experiments were then carried out.
Cell viability analysis
The cells were seeded into 96-well plate at 5×103/well and cultured in 10% FBS containing DMEM for 24h. Then, cells were treated in 100 µL of DMEM according to the above groups. After incubating for 24 h, add 10 µL of CCK-8 reagent to every well, and incubate for extra 4h. Absorbance of every well was measured at 450 nm. Each well has two duplicate wells to confirm the reproducibility.
Automated observation of axon growth with the live cell imager
Cells and axon growth trends were observed using Lionheart FX Automated Live Cell Imager (BioTeK, USA). The relevant treatment was given to every group following the study design. The control group was used to adjust the photo parameters. The duration of observation was 24 h, and data were recorded every hour.
Rotarod test
Mice were positioned on a Rotary rod instrument (UGO 47650, Italy) and skilled for 5 min at the 10 rpm speed for 3 days. For testing, mice were positioned on a rod that turned around accelerating velocity beginning with 5 rpm and accelerating up to 30 rpm in 60 s. Experiments were performed three times.
Forced running wheel test
Mice were positioned on a wheel fatigue meter (YLS-10B, Yiyan, China) to explore their staying power capabilities. Mice were skilled at 30 rpm for 10 min, and the fatigue running distance was determined. The wheel fatigue meter was stopped if mice fail to run within 10 sec after the electric shock (1 mA) and the wheel fatigue meter was started after mice rest for 30 sec. The experiments were performed in triplicates.
Open field test
The open field experimental analysis system for mice was provided by China Medical University. The mice were placed in the laboratory to adapt to the environment for 10 minutes. This experiment is performed in a quiet, low-light environment. The indoor temperature was maintained at about 26°C. The mice were positioned in the open field and the mice activity was observed within 5 min. Observation indicators include total horizontal movement distance and horizontal movement speed. Clean up the labyrinth after the experiment of each mouse, and try to avoid external interference that will cause unnecessary adverse effects on the experimental results.
Gait analysis
In order to enable all mice to walk to the end of the runway at a uniform speed without external stimulation, the mice were trained for 3 days. Data was collected on the 4th day and recorded 3 times for each mouse. The collected data was analyzed with Cat Walk XT Version10.6, and the average value of each index was taken. The related indexes are run duration, average speed, swing speed, and maximum variation.
Determination of Mn levels
The inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7700x, USA) was used for analysis of Mn levels in striatum of mice. Striatum was predigested, and 0.1, 1, 5, 10, 50, and 100µg/L series of Mn standard solutions were prepared for future use. The content of Mn in the striatum was then determined with internal standard method.
Measurement of DA levels
Total DA levels in the striatum were quantified using ELISA kits of mice DA (Baolai, Jiangsu, China) following the manufacturer's directions. The sample value within the detectable range was used for analysis. Experiments were performed in triplicates.
Immunofluorescence of FTO, ephrin-B2, TH, DARPP32, and DAT
Expression and localization of FTO and ephrin-B2 were determined in striatum and neurons, respectively, through immunofluorescence. In addition, immunofluorescence triple staining of TH, DARPP32 and DAT in striatum was used to explore whether nigro-striatal projection neurons were damaged. Samples were dehydrated with xylene and alcohol, and tissue sections were steamed with sodium citrate for antigen retrieval. Samples were blocked in 10% Goat serum for 30 min at 25°C. Samples were then treated with rabbit antibody of FTO (1:100), mouse antibody of ephrin-B2 (1:100), rabbit antibody of TH (1:100), mouse antibody of DARPP32 (1:50), and rat antibody of DAT (1:50) overnight at 4°C. On the second day, after 1 h of rewarming and washing 3 times with phosphate buffered solution (PBS), samples were treated with goat anti-mouse IgG (1:100), goat anti-rabbit IgG (1:100), and goat anti-rat IgG (1:100) for 1 h at 25°C. For immunofluorescence of FTO and ephrin-B2, samples were counterstained using 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) for 5 min after washing 3 times with PBS. Then, samples were mounted with mounting medium for further study. Laser scanning confocal microscope (A1R, Nikon, Japan) was used to capture images.
Hematoxylin and eosin (HE) staining
After the mice were perfused with 4% paraformaldehyde, the striatum tissue was removed. The striatum of mice was serially dehydrated in xylene and ethanol. The striatum was sectioned at 5 µm thickness after paraffin embedded. Then, HE staining was done for morphological observation with high resolution panoramic imaging system (CS2, Leica, Germany).
Golgi-cox staining
Brains of mice were removed quickly from the skull after euthanizing. After washing, the brains were stained with the FD Rapid GolgiStain™ kit (MD, USA). Firstly, the brains were incubated in the impregnation solution (A and B) for 14–15 days. Afterwards, the brains were treated with Solution C for the 48–72 h. Microtome Cryostat (1950, Leica, Germany) was used to cut 200 µm slices. Slices were mounted on a gelatin-coated slides, then dehydrated, stained, and coverslipped. Then, dendritic spines was observed with high resolution panoramic imaging system (CS2, Leica, Germany).
Nissl staining
Tissue sections were sequentially stained with series alcohol dehydration. The sections were then treated with 1% thionine for 1 h, then the samples were washed with distilled water. 70% alcohol color separation was performed for several sec to several minutes. The sections were dehydrated with 70%, 80%, 95%, and 100% alcohol, re-spectively, with 2 min for each. Next, the sections were washed with anhydrous ethanol twice (with 5 min for each time), followed by washing with xylene twice (with 10 min for each time). Finally, the sections were observed with high resolution panoramic imaging system (CS2, Leica, Germany).
Fluorogold retrograde tracing
Two mice in each group were injected with 2% FG solution into the striatum via brain stereotaxic injection. After 5 days of survival, the mice were anesthetized and perfused with 4% paraformaldehyde solution. The brain was taken out and fixed with 4% paraformaldehyde solution for 4 hours. Then the brain tissue was placed in a 20% sucrose solution at 4°C overnight. The next day, freeze sectioning was performed, and the section thickness was 30 µm. The sections were washed with 0.01 mM PBS and fixed in 4% paraformaldehyde for 10 min. After washing 3 times with PBS, samples were mounted with mounting medium for further study. Laser scanning confocal microscope (A1R, Nikon, Japan) was used to capture images.
Electron microscopy studies
After the mice were perfused with 4% paraformaldehyde, the striatum was fixed with 2.5% glutaraldehyde for 1h. A 1-mm-thick slice was made and fixed overnight. Postfixation of slice was carried out in 1% OsO4 for electron microscopy observation. Then, tissues were embedded in spur resin after dehydrated in series of acetone. Blocks were photographed in a transmission electron microscope (JEM-1400-FLASH, Japan) after stained with uranyl acetate and lead citrate.
Electrophysiological recording of striatum slices
Spontaneous discharge frequency in the striatum was recorded in 300 µm slices. Slices were cut using vibratome in a modified artificial cerebrospinal fluid (mACSF) saturated with 5% CO2/95% O2. Slices were then placed on an incubation chamber with normal ACSF and incubated at 25°C for at least 1 h before recording. Analysis was performed with a MED64 flat microelectrode array system (Alpha of Japan).
Western Blotting analysis
Total protein of striatum was extracted with RIPA buffer and protease inhibitors. BCA reagent was used for protein quantification. Proteins samples were transferred onto polyvinylidene difluoride (PVDF) membranes after gel electrophoresis. PVDF membranes were then blocked using 5% bovine serum albumin fraction V for 1h, then the membranes were incubated with primary antibody against FTO (1:500), METTL3 (1:1000), METTL14 (1:500), ALKBH5 (1:1000), YTHDF1 (1:2000), YTHDF2 (1:1000), ephrin-A1 (1:1000), ephrin-A2 (1:500), ephrin-A3 (1:1000), ephrin-A4 (1:500), ephrin-A5 (1:500), ephrin-B1 (1:1000), ephrin-B2 (1:1000), ephrin-B3 (1:500), and GAPDH (1:5000) overnight at 4°C. In the next day, membranes were incubated with corresponding secondary antibodies for 1 h at 25°C after washing 3 times with Tris-buffered saline with 0.1% Tween 20 (TBST). Analysis was performed with the multifunctional imaging system (AZURE C500, USA). GAPDH was used as an internal standard.
RT-qPCR assay
Total RNA was dissolved in RNase-free water (TaKaRa) after extraction using the Trizol reagent (TransGen, Beijing, China). First strand cDNA was obtained from total RNA using reverse transcriptase Kit (TaKaRa) following manufacturer’s instructions. RT-qPCR was performed using QuantStudio 6 Flex fluorescence quantitative PCR instrument (Thermo Fisher, USA). Primer sequences of FTO, METTL3, METTL14, ALKBH5, YTHDF1, YTHDF2, ephrin-A1, ephrin-A2, ephrin-A3, ephrin-A4, ephrin-A5, ephrin-B1, ephrin-B2, ephrin-B3, Grb4, GIT1, HPRT1, and β-actin for RT-qPCR analysis are presented in Supplementary Table 1. HPRT1 or β-actin genes were used as internal controls. Comparative CT method (ΔΔCT) was used to quantify the target genes. All experiments were performed in triplicates.
Quantifcation of m6A in total RNA
The ELISA-based m6A Quantitative Kit (Epigentek, P-9005-96) was used to quantify m6A levels of total RNA (200 ng). In accordance to the manufacturer’s instructions, quantifification was performed in triplicates.
Actinomycin D assay
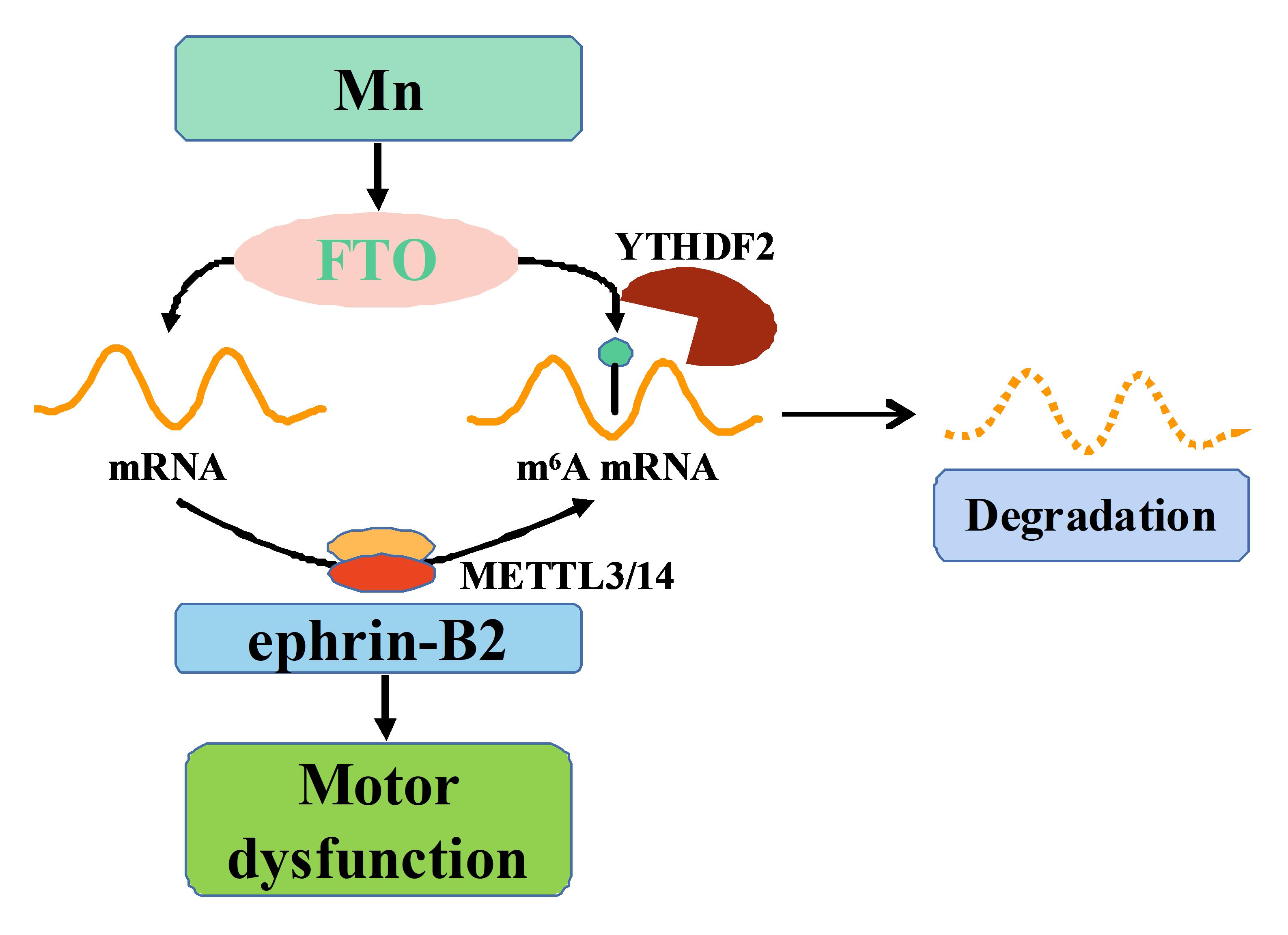
Samples were treated with 2µM actinomycin D (Act-D, Sigma, USA) for 0, 3, and 6 h. Cells were collected and RNA was isolated for RT-qPCR. HPRT1 gene was used as an internal control as HPRT1 mRNA does not bind to YTHDF2, is rarely affected by Act-D, and does not undergo m6A modifications.
m6A-RT-qPCR
Total RNA was used for m6A-immunoprecipitation and 500 ng of RNA was used as an input sample. 100 µg of RNA was diluted with 40 µl IP buffer (0.05 M Tris-HCl, 0.75 M NaCl, 0.5% Igepal, and RNase-free water) and treated with m6A antibody (Synaptic Systems, Germany) for 3.5 h at 4°C, Recombinant protein-A bead (Waltham, MA) containing BSA was added to the RNA solution and shaken at 4°C for 4 h. The m6A IP portion was eluted with elution buffer (300 µL 20mM CaCl2, 2µL RNasin, 2 µL RVC, and 0.75 µL 20 mg/mL Proteinase K) after washing three times with IP buffer. The collected total eluate was concentrated in an RNA concentrator to a final volume of 20 µL. The concentration of RNA was determined and reverse transcribed to cDNA. RT-qPCR was used to determine m6A levels of mRNA. Data were calculated using the %input = 2 (−ΔCtnormalized RIP) method.
Statistical analysis
Results were presented as means ± standard deviations after measured at least three times. The SPSS 22 software was used to analysis all statistical. The one-way analysis of variance (ANOVA) followed by Bonferroni test were used to determined the differences between the means for multiple comparison. P-value of < 0.05 or P-value of < 0.01 were considered statistically significant.
{kind=link}